-
2024 Sep 27
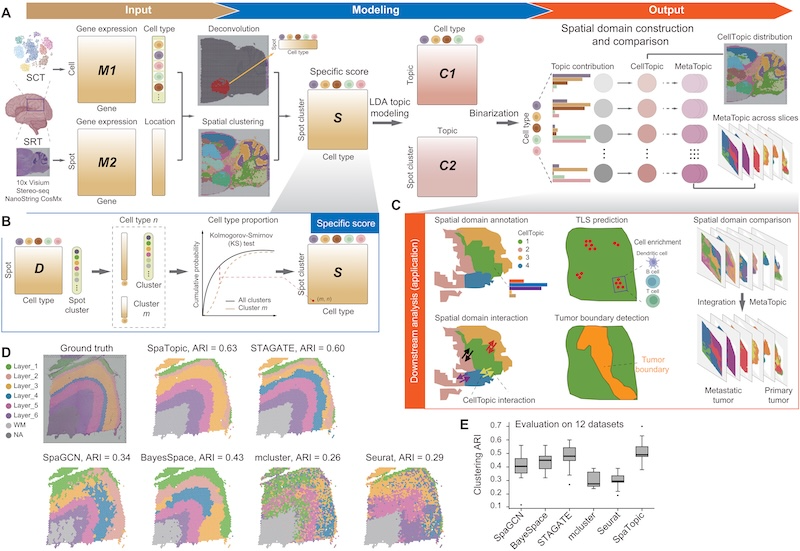 SpaTopic Spatial transcriptomic Spatial domains Tumor
SpaTopic Spatial transcriptomic Spatial domains Tumor74. SpaTopic: A statistical learning framework for exploring tumor spatial architecture from spatially resolved transcriptomic data.
Zhang Y, Yu B, Ming W, Zhou X, Wang J, Chen D*
Sci Adv. 2024 Sep 27;10(39):eadp4942. doi: 10.1126/sciadv.adp4942. Epub 2024 Sep 27.
Tumor tissues exhibit a complex spatial architecture within the tumor microenvironment (TME). Spatially resolved transcriptomics (SRT) is promising for unveiling the spatial structures of the TME at both cellular and molecular levels, but identifying pathology-relevant spatial domains remains challenging. Here, we introduce SpaTopic, a statistical learning framework that harmonizes spot clustering and cell-type deconvolution by integrating single-cell transcriptomics and SRT data. Through topic modeling, SpaTopic stratifies the TME into spatial domains with coherent cellular organization, facilitating refined annotation of the spatial architecture with improved performance. We assess SpaTopic across various tumor types and show accurate prediction of tertiary lymphoid structures and tumor boundaries. Moreover, marker genes derived from SpaTopic are transferrable and can be applied to mark spatial domains in other datasets. In addition, SpaTopic enables quantitative comparison and functional characterization of spatial domains across SRT datasets. Overall, SpaTopic presents an innovative analytical framework for exploring, comparing, and interpreting tumor SRT data.
-
2024 Aug 3
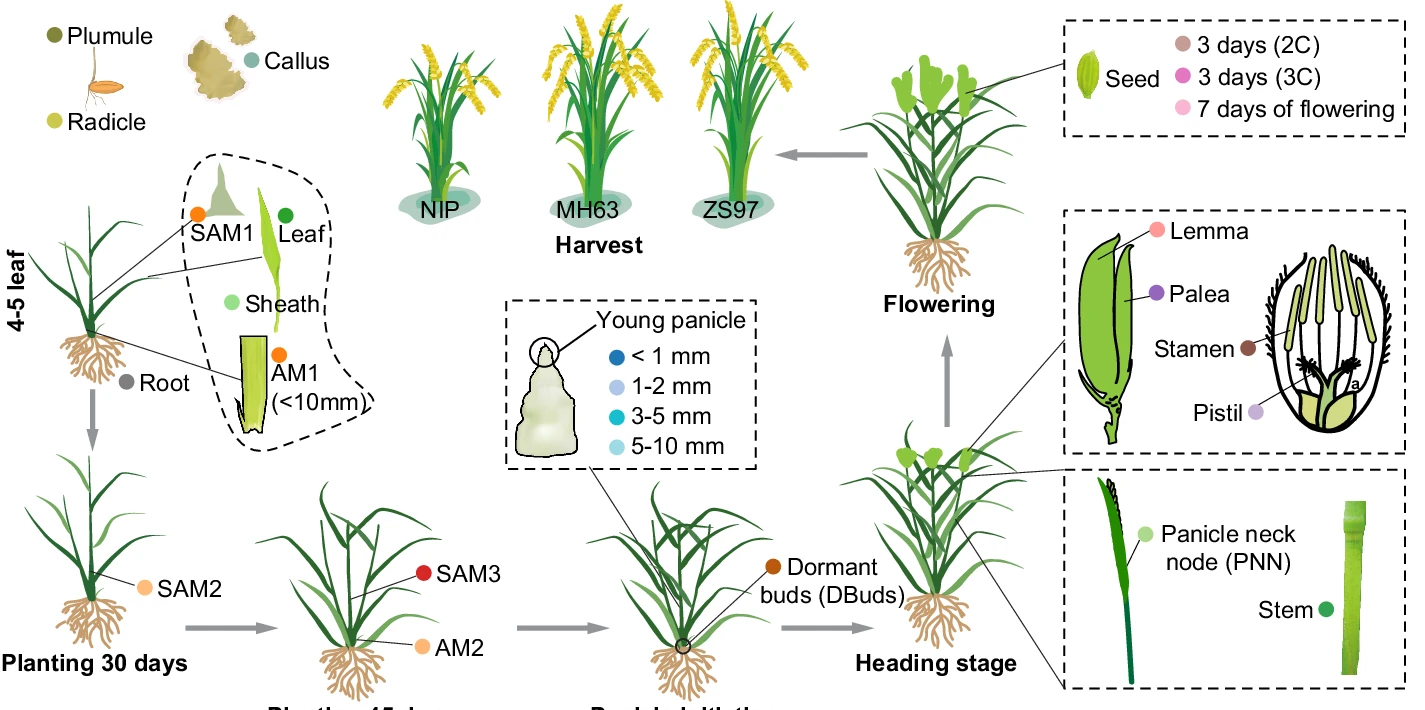 ATAC-seq Chromatin accessibility Regulome GWAS Rice
ATAC-seq Chromatin accessibility Regulome GWAS Rice73. Comprehensive mapping and modelling of the rice regulome landscape unveils the regulatory architecture underlying complex traits.
Zhu T#, Xia C#, Yu R, Zhou X, Xu X, Wang L, Zong Z, Yang J, Liu Y, Ming L, You Y, Chen D*, Xie W
Nat Commun. 2024 Aug 3;15(1):6562. doi: 10.1038/s41467-024-50787-y.
Unraveling the regulatory mechanisms that govern complex traits is pivotal for advancing crop improvement. Here we present a comprehensive regulome atlas for rice (Oryza sativa), charting the chromatin accessibility across 23 distinct tissues from three representative varieties. Our study uncovers 117,176 unique open chromatin regions (OCRs), accounting for ~15% of the rice genome, a notably higher proportion compared to previous reports in plants. Integrating RNA-seq data from matched tissues, we confidently predict 59,075 OCR-to-gene links, with enhancers constituting 69.54% of these associations, including many known enhancer-to-gene links. Leveraging this resource, we re-evaluate genome-wide association study results and discover a previously unknown function of OsbZIP06 in seed germination, which we subsequently confirm through experimental validation. We optimize deep learning models to decode regulatory grammar, achieving robust modeling of tissue-specific chromatin accessibility. This approach allows to predict cross-variety regulatory dynamics from genomic sequences, shedding light on the genetic underpinnings of cis-regulatory divergence and morphological disparities between varieties. Overall, our study establishes a foundational resource for rice functional genomics and precision molecular breeding, providing valuable insights into regulatory mechanisms governing complex traits.
doi: 10.1038/s41467-024-50787-y PubMed: 39095348 Google Scholar
-
2023 Aug 23
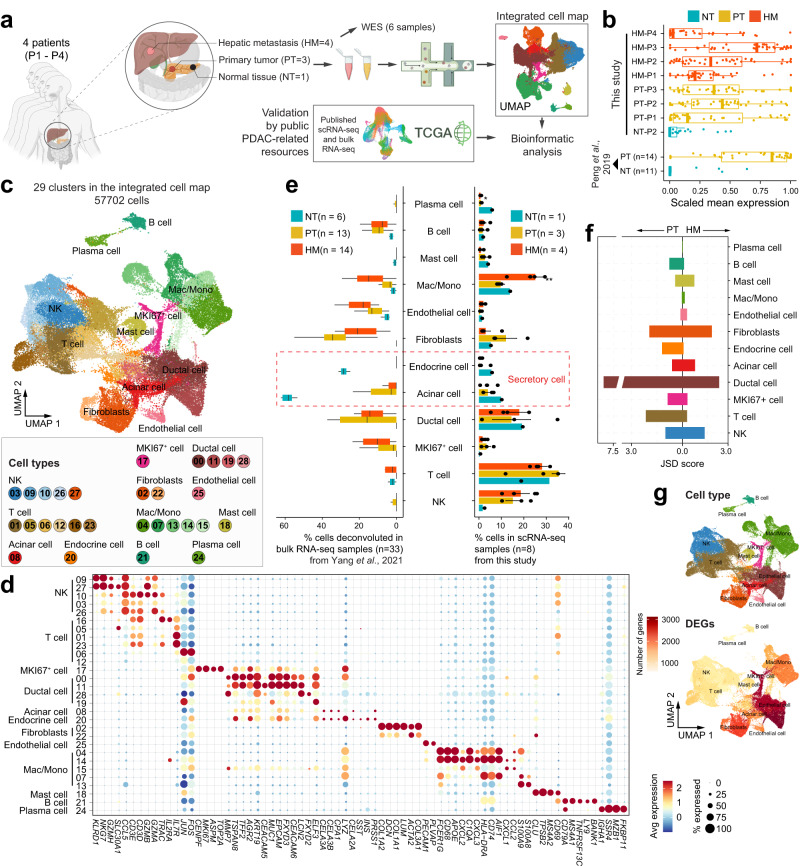 Tumor microenvironment Metastasis Pancreatic cancer
Tumor microenvironment Metastasis Pancreatic cancer65. Single cell transcriptomic analyses implicate an immunosuppressive tumor microenvironment in pancreatic cancer liver metastasis.
Zhang S#, Fang W#, Zhou S#, Zhu D#, Chen R#, Gao X, Li Z, Fu Y, Zhang Y, Yang F, Zhao J, Wu H, Wang P, Shen Y, Shen S, Xu G, Wang L, Yan C, Zou X, Chen D*, Lv Y
Nat Commun. 2023 Aug 23;14(1):5123. doi: 10.1038/s41467-023-40727-7.
Pancreatic ductal adenocarcinoma (PDAC) is a highly metastatic disease refractory to all targeted and immune therapies. However, our understanding of PDAC microenvironment especially the metastatic microenvironment is very limited partly due to the inaccessibility to metastatic tumor tissues. Here, we present the single-cell transcriptomic landscape of synchronously resected PDAC primary tumors and matched liver metastases. We perform comparative analysis on both cellular composition and functional phenotype between primary and metastatic tumors. Tumor cells exhibit distinct transcriptomic profile in liver metastasis with clearly defined evolutionary routes from cancer cells in primary tumor. We also identify specific subtypes of stromal and immune cells critical to the formation of the pro-tumor microenvironment in metastatic lesions, including RGS5+ cancer-associated fibroblasts, CCL18+ lipid-associated macrophages, S100A8+ neutrophils and FOXP3+ regulatory T cells. Cellular interactome analysis further reveals that the lack of tumor-immune cell interaction in metastatic tissues contributes to the formation of the immunosuppressive microenvironment. Our study provides a comprehensive characterization of the transcriptional landscape of PDAC liver metastasis.
doi: 10.1038/s41467-023-40727-7 PubMed: 37612267 Google Scholar
-
2022 Jul 13
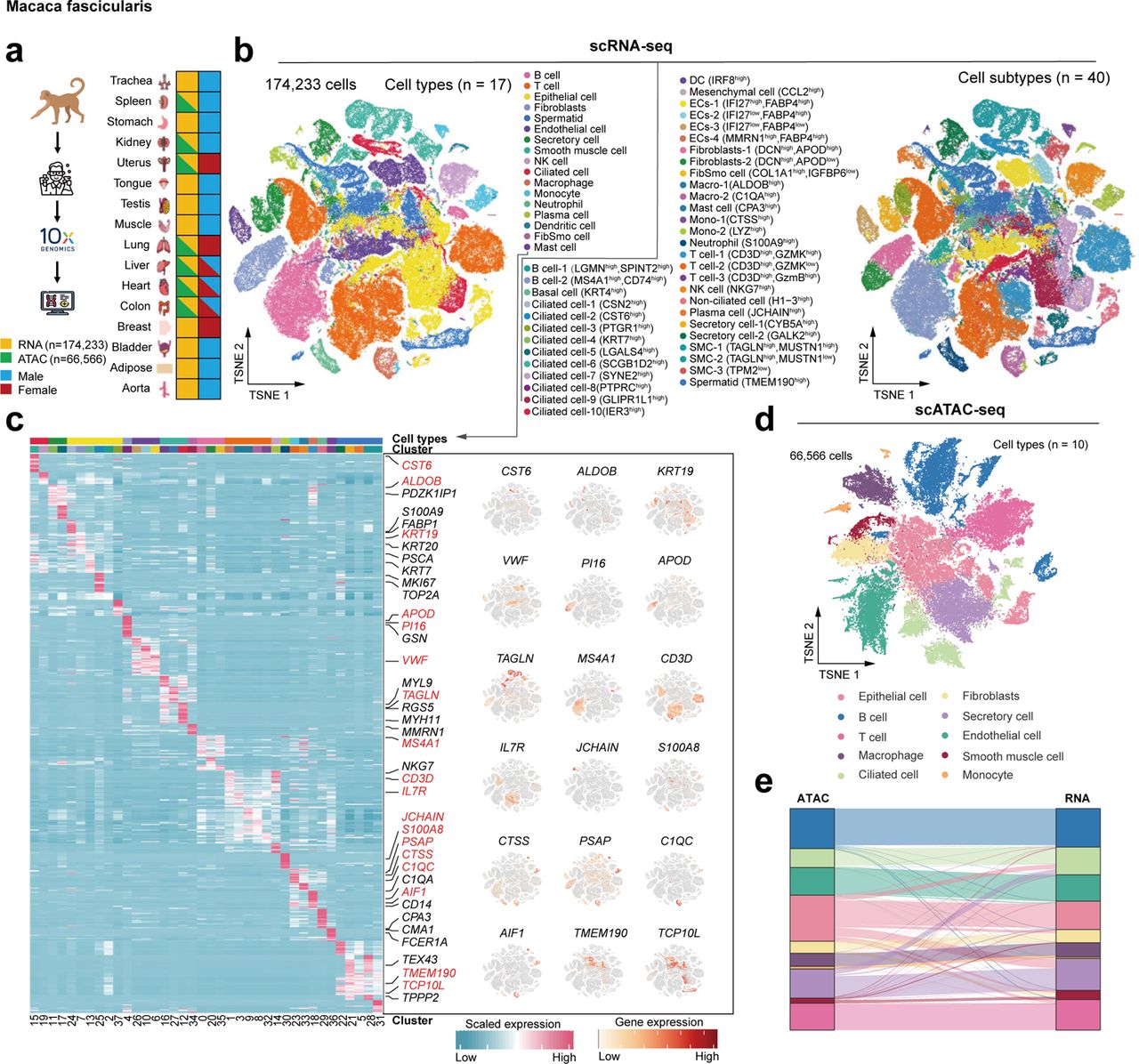 Cynomolgus monkey Cell atlas scRNA-seq scATAC-seq
Cynomolgus monkey Cell atlas scRNA-seq scATAC-seq56. A reference single-cell regulomic and transcriptomic map of cynomolgus monkeys.
Qu J#, Yang F#, Zhu T#, Wang Y#, Fang W, Ding Y, Zhao X, Qi X, Xie Q, Chen M, Xu Q, Xie Y, Sun Y, Chen D*
Nat Commun. 2022 Jul 13;13(1):4069. doi: 10.1038/s41467-022-31770-x.
Non-human primates are attractive laboratory animal models that accurately reflect both developmental and pathological features of humans. Here we present a compendium of cell types across multiple organs in cynomolgus monkeys (Macaca fascicularis) using both single-cell chromatin accessibility and RNA sequencing data. The integrated cell map enables in-depth dissection and comparison of molecular dynamics, cell-type compositions and cellular heterogeneity across multiple tissues and organs. Using single-cell transcriptomic data, we infer pseudotime cell trajectories and cell-cell communications to uncover key molecular signatures underlying their cellular processes. Furthermore, we identify various cell-specific cis-regulatory elements and construct organ-specific gene regulatory networks at the single-cell level. Finally, we perform comparative analyses of single-cell landscapes among mouse, monkey and human. We show that cynomolgus monkey has strikingly higher degree of similarities in terms of immune-associated gene expression patterns and cellular communications to human than mouse. Taken together, our study provides a valuable resource for non-human primate cell biology.
doi: 10.1038/s41467-022-31770-x PubMed: 35831300 Google Scholar
-
2022 Jun 14
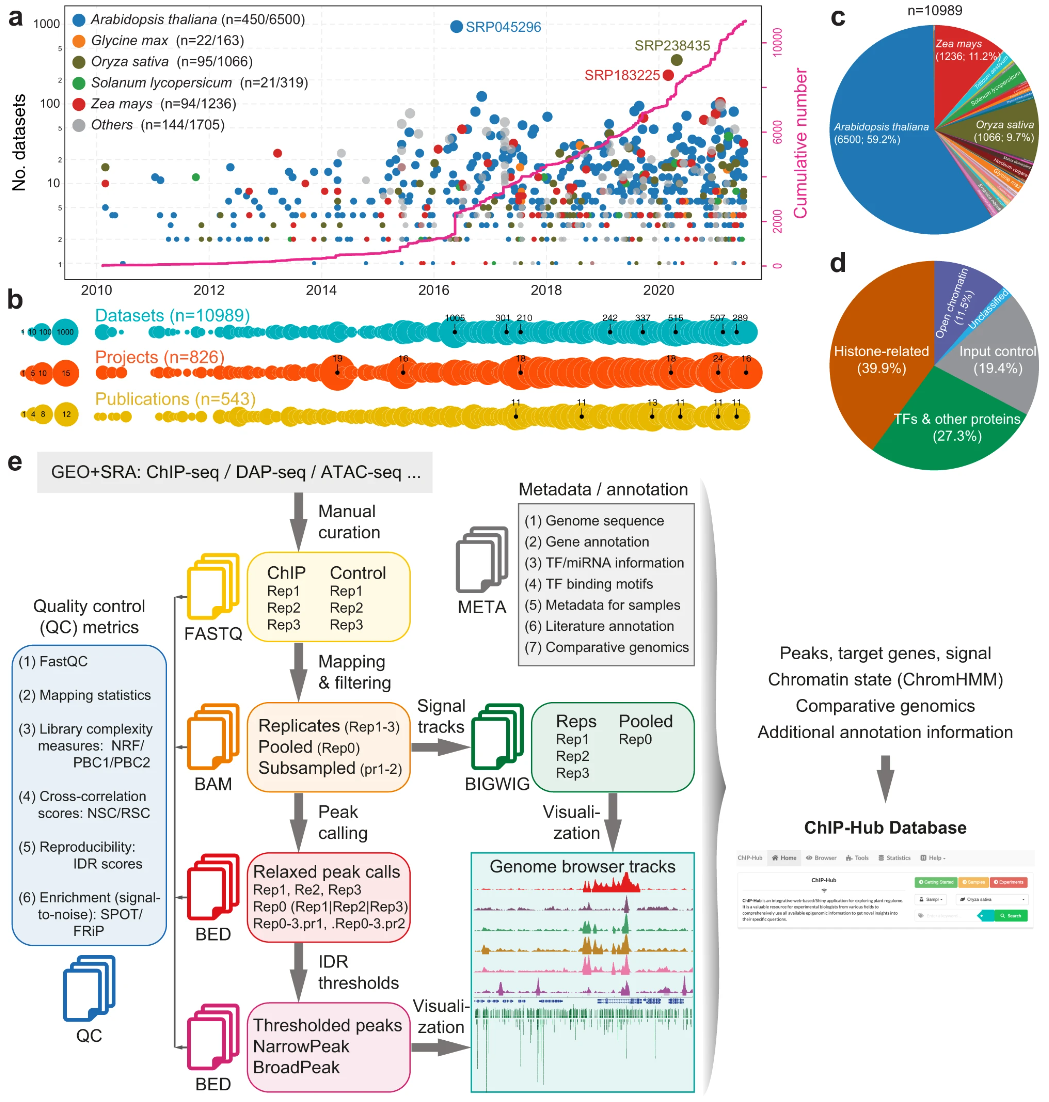 ChIP-Hub ATAC-seq ChIP-seq Regulome Plants
ChIP-Hub ATAC-seq ChIP-seq Regulome Plants55. ChIP-Hub provides an integrative platform for exploring plant regulome.
Fu LY#, Zhu T#, Zhou X#, Yu R#, He Z, Zhang P, Wu Z, Chen M, Kaufmann K, Chen D*
Nat Commun. 2022 Jun 14;13(1):3413. doi: 10.1038/s41467-022-30770-1.
Plant genomes encode a complex and evolutionary diverse regulatory grammar that forms the basis for most life on earth. A wealth of regulome and epigenome data have been generated in various plant species, but no common, standardized resource is available so far for biologists. Here, we present ChIP-Hub, an integrative web-based platform in the ENCODE standards that bundles >10,000 publicly available datasets reanalyzed from >40 plant species, allowing visualization and meta-analysis. We manually curate the datasets through assessing ~540 original publications and comprehensively evaluate their data quality. As a proof of concept, we extensively survey the co-association of different regulators and construct a hierarchical regulatory network under a broad developmental context. Furthermore, we show how our annotation allows to investigate the dynamic activity of tissue-specific regulatory elements (promoters and enhancers) and their underlying sequence grammar. Finally, we analyze the function and conservation of tissue-specific promoters, enhancers and chromatin states using comparative genomics approaches. Taken together, the ChIP-Hub platform and the analysis results provide rich resources for deep exploration of plant ENCODE. ChIP-Hub is available at https://biobigdata.nju.edu.cn/ChIPHub/ .
doi: 10.1038/s41467-022-30770-1 PubMed: 35701419 Google Scholar
-
2019 Apr 12
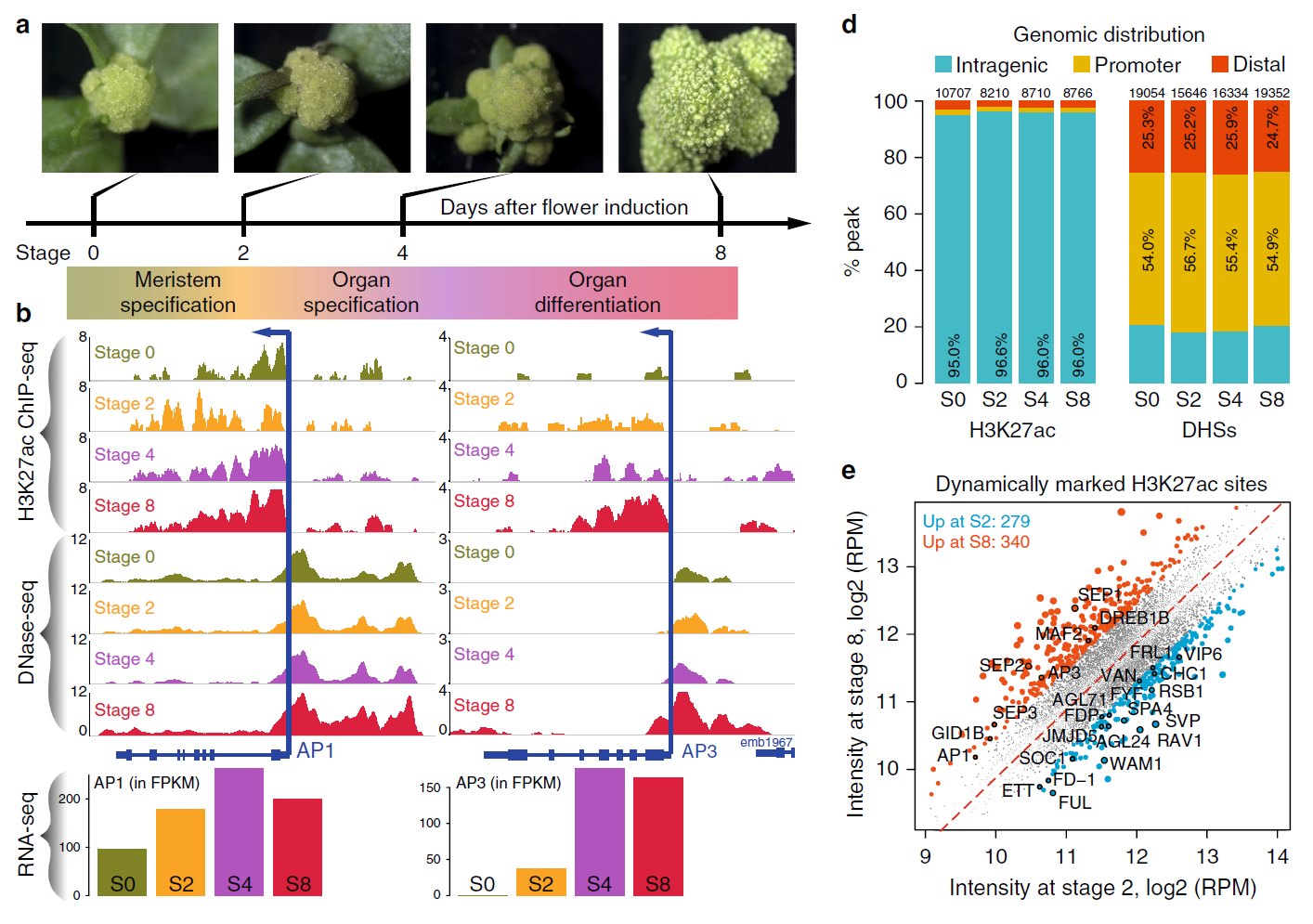 Enhancer H3K27ac DNase І hypersensitivity sites (DHSs) DNase-seq ChIP-seq RNA-seq Transcription factor (TF) Arabidopsis thaliana Plant
Enhancer H3K27ac DNase І hypersensitivity sites (DHSs) DNase-seq ChIP-seq RNA-seq Transcription factor (TF) Arabidopsis thaliana Plant42. Dynamic control of enhancer activity drives stage-specific gene expression during flower morphogenesis.
Yan W, Chen D#,*, Schumacher J, Durantini D, Engelhorn J, Chen M, Carles CC, Kaufmann K
Nat Commun. 2019 Apr 12;10(1):1705. doi: 10.1038/s41467-019-09513-2.
Enhancers are critical for developmental stage-specific gene expression, but their dynamic regulation in plants remains poorly understood. Here we compare genome-wide localization of H3K27ac, chromatin accessibility and transcriptomic changes during flower development in Arabidopsis. H3K27ac prevalently marks promoter-proximal regions, suggesting that H3K27ac is not a hallmark for enhancers in Arabidopsis. We provide computational and experimental evidence to confirm that distal DNase І hypersensitive sites are predictive of enhancers. The predicted enhancers are highly stage-specific across flower development, significantly associated with SNPs for flowering-related phenotypes, and conserved across crucifer species. Through the integration of genome-wide transcription factor (TF) binding datasets, we find that floral master regulators and stage-specific TFs are largely enriched at developmentally dynamic enhancers. Finally, we show that enhancer clusters and intronic enhancers significantly associate with stage-specific gene regulation by floral master TFs. Our study provides insights into the functional flexibility of enhancers during plant development, as well as hints to annotate plant enhancers.
doi: 10.1038/s41467-019-09513-2 PubMed: 30979870 Google Scholar
-
2018 Oct 31
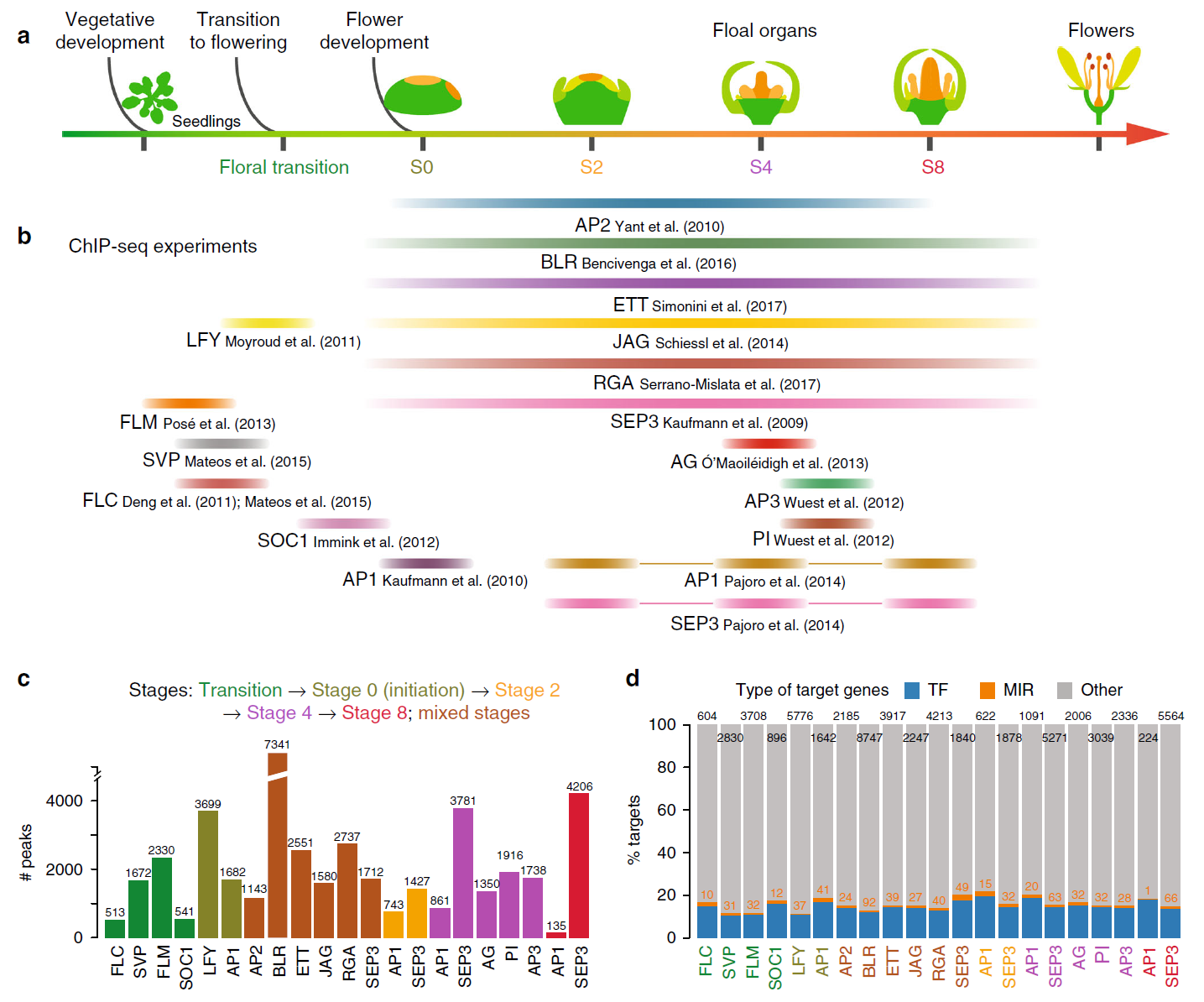 Flower development Gene regulatory networks (GRNs) Transcription factors (TFs) microRNAs (miRNAs) Feed-forward loops (FFLs) Arabidopsis thaliana Plant
Flower development Gene regulatory networks (GRNs) Transcription factors (TFs) microRNAs (miRNAs) Feed-forward loops (FFLs) Arabidopsis thaliana Plant41. Architecture of gene regulatory networks controlling flower development in Arabidopsis thaliana.
Chen D*, Yan W, Fu LY, Kaufmann K
Nat Commun. 2018 Oct 31;9(1):4534. doi: 10.1038/s41467-018-06772-3.
Floral homeotic transcription factors (TFs) act in a combinatorial manner to specify the organ identities in the flower. However, the architecture and the function of the gene regulatory network (GRN) controlling floral organ specification is still poorly understood. In particular, the interconnections of homeotic TFs, microRNAs (miRNAs) and other factors controlling organ initiation and growth have not been studied systematically so far. Here, using a combination of genome-wide TF binding, mRNA and miRNA expression data, we reconstruct the dynamic GRN controlling floral meristem development and organ differentiation. We identify prevalent feed-forward loops (FFLs) mediated by floral homeotic TFs and miRNAs that regulate common targets. Experimental validation of a coherent FFL shows that petal size is controlled by the SEPALLATA3-regulated miR319/TCP4 module. We further show that combinatorial DNA-binding of homeotic factors and selected other TFs is predictive of organ-specific patterns of gene expression. Our results provide a valuable resource for studying molecular regulatory processes underlying floral organ specification in plants.
doi: 10.1038/s41467-018-06772-3 PubMed: 30382087 Google Scholar
-
2014 Dec 11
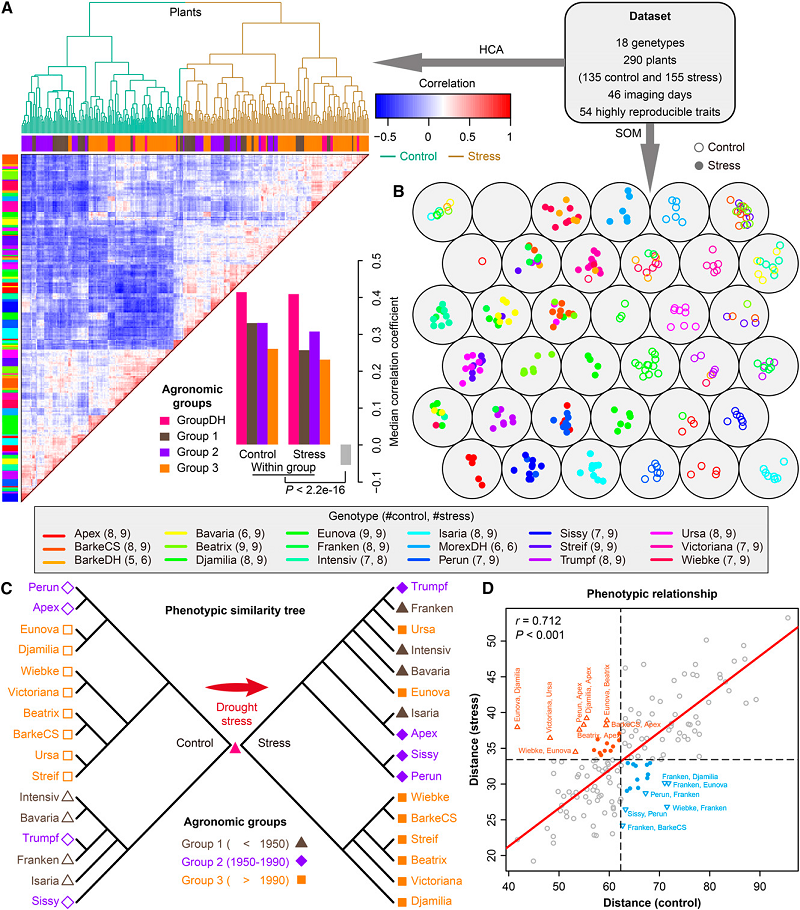 High-throughput image analysis Plant phenotyping Phenomics Hordeum vulgare (barley) Plant
High-throughput image analysis Plant phenotyping Phenomics Hordeum vulgare (barley) Plant23. Dissecting the phenotypic components of crop plant growth and drought responses based on high-throughput image analysis.
Chen D, Neumann K, Friedel S, Kilian B, Chen M, Altmann T, Klukas C
Plant Cell. 2014 Dec;26(12):4636-55. doi: 10.1105/tpc.114.129601.
Significantly improved crop varieties are urgently needed to feed the rapidly growing human population under changing climates. While genome sequence information and excellent genomic tools are in place for major crop species, the systematic quantification of phenotypic traits or components thereof in a high-throughput fashion remains an enormous challenge. In order to help bridge the genotype to phenotype gap, we developed a comprehensive framework for high-throughput phenotype data analysis in plants, which enables the extraction of an extensive list of phenotypic traits from nondestructive plant imaging over time. As a proof of concept, we investigated the phenotypic components of the drought responses of 18 different barley (Hordeum vulgare) cultivars during vegetative growth. We analyzed dynamic properties of trait expression over growth time based on 54 representative phenotypic features. The data are highly valuable to understand plant development and to further quantify growth and crop performance features. We tested various growth models to predict plant biomass accumulation and identified several relevant parameters that support biological interpretation of plant growth and stress tolerance. These image-based traits and model-derived parameters are promising for subsequent genetic mapping to uncover the genetic basis of complex agronomic traits. Taken together, we anticipate that the analytical framework and analysis results presented here will be useful to advance our views of phenotypic trait components underlying plant development and their responses to environmental cues.
See also: Google Scholar - Pubmed
Jump to year: 2009 / 2010 / 2011 / 2012 / 2013 / 2014 / 2015 / 2016 / 2017 / 2018 / 2019 / 2020 / 2021 / 2022 / 2023 / 2024 / 2025
2025

84. Genetic effects on chromatin accessibility reveal the molecular mechanisms of complex traits in maize.
Zhu Y, Ngan H, Liu W, Zhu T, Li W, Xiao Y, Zhuo L, Chen D, Tu X, Gao K, Yan J, Zhong S, Yang N
Plant J. 2025 Aug;123(4):e70437. doi: 10.1111/tpj.70437.
Cis-regulatory elements (CREs) are critical for modulating gene expression and phenotypic diversity in maize. While genome-wide association study (GWAS) hits and expression quantitative trait loci (eQTLs) are often enriched in CREs, their molecular mechanisms remain poorly understood. Characterizing CREs within accessible chromatin regions (ACRs) offers a powerful approach to link noncoding variants to chromatin structure alterations and phenotypic variation. Here, we generated ATAC-seq profiles from seedling leaves of 214 maize inbred lines, identifying 82 174 consensus ACRs. Notably, 39.55% of these ACRs exhibited significant population-wide chromatin accessibility variation. By mapping chromatin accessibility quantitative trait loci (caQTLs), we discovered 27 004 loci, including 1398 predicted to disrupt transcription factor (TF)-binding sites. Integration with multi-omics data revealed 7405 caACR-target gene pairs and linked 56 caACRs to GWAS signals for 51 agronomic traits, with significant enrichment in flowering-related pathways. Functional candidates such as ZmZIM30 - putatively regulated by caACRs - emerged as key regulators of flowering time. At the fad7 locus associated with linolenic acid content, allelic variants overlapping a caQTL showed differential chromatin accessibility. Our study provides a high-resolution cis-elements of maize leaves, deciphers the genetic basis of chromatin accessibility variation, and bridges noncoding caQTLs to molecular mechanisms underlying GWAS hits.

83. The histone acetyltransferase GCN5 regulates floral meristem activity and flower development in Arabidopsis.
Hawar A, Chen W, Zhu T, Wang X, Liu J, Xiong S, Ito T, Chen D*, Sun B
Plant Cell. 2025 Jun 4;37(6):koaf135. doi: 10.1093/plcell/koaf135.
The histone acetyltransferase (HAT) GENERAL CONTROL NON DEREPRESSIBLE5 (GCN5) participates in various developmental processes in Arabidopsis (Arabidopsis thaliana). Notably, GCN5 ensures proper flower development, but the underlying mechanism remains unknown. Here, we show that during early flower development, GCN5 catalyzes histone acetylation at WUSCHEL (WUS) and CLAVATA3 (CLV3) chromatin, activating their expression. WUS and CLV3 are required for floral meristem (FM) maintenance. Furthermore, we demonstrate that the GCN5-ALTERATION/DEFICIENCY IN ACTIVATION 2 (ADA2) HAT module interacts with the Switch/Sucrose non-fermentable ATPase SPLAYED (SYD) to form a GCN5-ADA2b-SYD ternary complex. The cytokinin-responsive type-B Arabidopsis response regulators recruit this ternary complex for WUS activation. During floral organogenesis, PERIANTHIA recruits the GCN5-ADA2b module for AGAMOUS activation, which promotes FM determinacy. GCN5 also activates KNUCKLES, which ensures the timely termination of FM activity. Moreover, GCN5 modulates the expression pattern of the B-class gene APETALA3 and promotes the expression of SUPERMAN and CRABS CLAW, which are required for FM determinacy, thereby safeguarding meristem determinacy and correct floral organ formation. Thus, our study demonstrates the indispensable role of GCN5 in establishing a permissive chromatin environment to regulate the key genes required for precise flower development.

82. Integration of metabolome and transcriptome reveals flavonoid metabolic network during the developmental stage of flower buds in Cornus wilsoniana
Cai J, Chao H, Chen M, Chen D*
Med Plant Biol 4: e017 doi: 10.48130/mpb-0025-0015
Secondary metabolites (SMs) are crucial for plant adaptation and human health, yet the regulatory mechanisms underlying their biosynthesis remain incompletely understood. Cornus wilsoniana, a woody oil crop and medicinal plant, is renowned for its rich flavonoid content, yet the genetic and metabolic basis of its secondary metabolism remains largely unexplored. Here, we integrate transcriptomic and metabolomic analyses to dissect the regulatory landscape governing flavonoid biosynthesis during flower bud development. Comparative analyses between high-yield and low-yield genotypes across distinct developmental stages reveal a coordinated reprogramming of flavonoid pathways, with significant shifts in gene expression and metabolite accumulation. WGCNA links critical biosynthetic genes to co-expression modules enriched in hormone signaling, redox homeostasis, and light perception, underscoring the interplay between developmental and environmental cues in shaping metabolic fluxes. Furthermore, comparative genomics reveals 17 candidate genes forming a complete anthocyanin and proanthocyanidin biosynthetic network. Our findings illuminate the molecular framework underlying flavonoid metabolism in C. wilsoniana, providing a foundation for metabolic engineering and genetic improvement strategies aimed at enhancing bioactive compound production for medicinal and agricultural applications.

81. PGCP: A comprehensive database of plant genomes for comparative phylogenomics.
Zhou X#, Fan HY#, Feng XY#, Ruan Z#, Yuan J, Han Q, He Z, You Y, Chao H, Chen M, Shao ZQ, Xue JY, Chen D*
Plant Biotechnol J. 2025 Jul;23(7):2928-2930. doi: 10.1111/pbi.70110. Epub 2025 May 6.

80. Harnessing the Foundation Model for Exploration of Single-cell Expression Atlases in Plants.
Cao G, Chao H, Zheng W, Lan Y, Lu K, Wang Y, Chen M, Zhang H, Chen D*
Genomics Proteomics Bioinformatics. 2025 Mar 17:qzaf024. doi: 10.1093/gpbjnl/qzaf024. Online ahead of print.
Single-cell RNA sequencing (scRNA-seq) provides unprecedented insights into plant cellular diversity by enabling high-resolution analyses of gene expression at the single-cell level. However, the complexity of scRNA-seq data, including challenges in batch integration, cell type annotation, and gene regulatory network (GRN) inference, demands advanced computational approaches. To address these challenges, we developed scPlantLLM, a Transformer model trained on millions of plant single-cell data points. Using a sequential pretraining strategy incorporating masked language modeling and cell type annotation tasks, scPlantLLM generates robust and interpretable single-cell data embeddings. When applied to Arabidopsis thaliana datasets, scPlantLLM excels in clustering, cell type annotation, and batch integration, achieving an accuracy of up to 0.91 in zero-shot learning scenarios. Furthermore, the model demonstrates an ability to identify biologically meaningful GRNs and subtle cellular subtypes, showcasing its potential to advance plant biology research. Compared to traditional methods, scPlantLLM outperforms in key metrics such as adjusted rand index (ARI), normalized mutual information (NMI) and silhouette score (SIL), highlighting its superior clustering accuracy and biological relevance. scPlantLLM represents a foundational model for exploring plant single-cell expression atlases, offering unprecedented capabilities to resolve cellular heterogeneity and regulatory dynamics across diverse plant systems. The code used in this study is available at https://github.com/compbioNJU/scPlantLLM.

79. MUSIC-GCN: A Novel Multi-Tasking Pipeline for Analyzing Single-Cell Transcriptomic Data Using Residual Graph Convolution Network
Liu Y, Wei G, Li C, Shen LC, Gasser RB, Song J, Chen D*, Yu DJ
IEEE Transactions on Computational Biology and Bioinformatics, vol. 22, no. 2, pp. 781-789, March-April 2025
Single-cell transcriptomics is a powerful approach for characterizing gene transcription at cellular resolution. This approach requires efficient computational pipelines to undertake essential tasks, including clustering, dimensionality reduction, imputation, and denoising. Currently, most such pipelines undertake these computational tasks separately without considering the interdependence among these tasks. Here, we present an advanced pipeline, MUSIC-GCN, by employing a graph convolutional neural (GCN) network and autoencoder to perform multi-task single-cell RNA-sequencing (scRNA-seq) data analysis. The rationale is that multiple related tasks can be carried out simultaneously to enable enhanced learning and more effective representations through the ‘sharing of knowledge’ regarding individual tasks. Benchmarking experiments using various scRNA-seq datasets show that MUSIC-GCN can achieve a competitive performance on multi-tasks when benchmarked with state-of-the-art approaches.
2024

78. Monkey multi-organ cell atlas exposed to estrogen.
Fang W, Qu J, Zhao W, Cao X, Liu J, Han Q, Chen D, Lv W, Xie Y, Sun Y
Life Med. 2024 Mar 22;3(2):lnae012. doi: 10.1093/lifemedi/lnae012. eCollection 2024 Apr.
Awareness of estrogen's effects on health is broadening rapidly. The effects of long-term high levels of estrogen on the body involve multiple organs. Here, we used both single-cell chromatin accessibility and RNA sequencing data to analyze the potential effect of estrogen on major organs. The integrated cell map enabled in-depth dissection and comparison of molecular dynamics, cell-type compositions, and cellular heterogeneity across multiple tissues and organs under estrogen stimulation. We also inferred pseudotime cell trajectories and cell-cell communications to uncover key molecular signatures underlying their cellular processes in major organs in response to estrogen. For example, estrogen could induce the differentiation of IFIT3 + neutrophils into S100A9 + neutrophils involved in the function of endosome-to-lysosome transport and the multivesicular body sorting pathway in liver tissues. Furthermore, through integration with human genome-wide association study data, we further identified a subset of risk genes during disease development that were induced by estrogen, such as AKT1 (related to endometrial cancer), CCND1 (related to breast cancer), HSPH1 (related to colorectal cancer), and COVID-19 and asthma-related risk genes. Our work uncovers the impact of estrogen on the major organs, constitutes a useful resource, and reveals the contribution and mechanism of estrogen to related diseases.
doi: 10.1093/lifemedi/lnae012 PubMed: 39872660 Google Scholar

77. Deep learning on chromatin profiles reveals the cis-regulatory sequence code of the rice genome.
Zhou X, Ruan Z, Zhang C, Kaufmann K, Chen D*
J Genet Genomics. 2025 Jun;52(6):848-851. doi: 10.1016/j.jgg.2024.12.007. Epub 2024 Dec 18.

76. Non-coding RNA notations, regulations and interactive resources.
Cheng M, Zhu Y, Yu H, Shao L, Zhang Y, Li L, Tu H, Xie L, Chao H, Zhang P, Xin S, Feng C, Ivanisenko V, Orlov Y, Chen D, Wong A, Yang YE, Chen M
Funct Integr Genomics. 2024 Nov 18;24(6):217. doi: 10.1007/s10142-024-01494-w.
An increasing number of non-coding RNAs (ncRNAs) are found to have roles in gene expression and cellular regulations. However, there are still a large number of ncRNAs whose functions remain to be studied. Despite decades of research, the field continues to evolve, with each newly identified ncRNA undergoing processes such as biogenesis, identification, and functional annotation. Bioinformatics methodologies, alongside traditional biochemical experimental methods, have played an important role in advancing ncRNA research across various stages. Presently, over 50 types of ncRNAs have been characterized, each exhibiting diverse functions. However, there remains a need for standardization and integration of these ncRNAs within a unified framework. In response to this gap, this review traces the historical trajectory of ncRNA research and proposes a unified notation system. Additionally, we comprehensively elucidate the ncRNA interactome, detailing its associations with DNAs, RNAs, proteins, complexes, and chromatin. A web portal named ncRNA Hub ( https://bis.zju.edu.cn/nchub/ ) is also constructed to provide detailed notations of ncRNAs and share a collection of bioinformatics resources. This review aims to provide a broader perspective and standardized paradigm for advancing ncRNA research.
doi: 10.1007/s10142-024-01494-w PubMed: 39557706 Google Scholar

75. iSeq: an integrated tool to fetch public sequencing data.
Chao H, Li Z, Chen D*, Chen M
Bioinformatics. 2024 Nov 1;40(11):btae641. doi: 10.1093/bioinformatics/btae641.
MOTIVATION: High-throughput sequencing technologies [next-generation sequencing (NGS)] are increasingly used to address diverse biological questions. Despite the rich information in NGS data, particularly with the growing datasets from repositories like the Genome Sequence Archive (GSA) at NGDC, programmatic access to public sequencing data and metadata remains limited. RESULTS: We developed iSeq to enable quick and straightforward retrieval of metadata and NGS data from multiple databases via the command-line interface. iSeq supports simultaneous retrieval from GSA, SRA, ENA, and DDBJ databases. It handles over 25 different accession formats, supports Aspera downloads, parallel downloads, multi-threaded processes, FASTQ file merging, and integrity verification, simplifying data acquisition and enhancing the capacity for reanalyzing NGS data. AVAILABILITY AND IMPLEMENTATION: iSeq is freely available on Bioconda (https://anaconda.org/bioconda/iseq) and GitHub (https://github.com/BioOmics/iSeq).
doi: 10.1093/bioinformatics/btae641 PubMed: 39447029 Google Scholar

74. SpaTopic: A statistical learning framework for exploring tumor spatial architecture from spatially resolved transcriptomic data.
Zhang Y, Yu B, Ming W, Zhou X, Wang J, Chen D*
Sci Adv. 2024 Sep 27;10(39):eadp4942. doi: 10.1126/sciadv.adp4942. Epub 2024 Sep 27.
Tumor tissues exhibit a complex spatial architecture within the tumor microenvironment (TME). Spatially resolved transcriptomics (SRT) is promising for unveiling the spatial structures of the TME at both cellular and molecular levels, but identifying pathology-relevant spatial domains remains challenging. Here, we introduce SpaTopic, a statistical learning framework that harmonizes spot clustering and cell-type deconvolution by integrating single-cell transcriptomics and SRT data. Through topic modeling, SpaTopic stratifies the TME into spatial domains with coherent cellular organization, facilitating refined annotation of the spatial architecture with improved performance. We assess SpaTopic across various tumor types and show accurate prediction of tertiary lymphoid structures and tumor boundaries. Moreover, marker genes derived from SpaTopic are transferrable and can be applied to mark spatial domains in other datasets. In addition, SpaTopic enables quantitative comparison and functional characterization of spatial domains across SRT datasets. Overall, SpaTopic presents an innovative analytical framework for exploring, comparing, and interpreting tumor SRT data.

73. Comprehensive mapping and modelling of the rice regulome landscape unveils the regulatory architecture underlying complex traits.
Zhu T#, Xia C#, Yu R, Zhou X, Xu X, Wang L, Zong Z, Yang J, Liu Y, Ming L, You Y, Chen D*, Xie W
Nat Commun. 2024 Aug 3;15(1):6562. doi: 10.1038/s41467-024-50787-y.
Unraveling the regulatory mechanisms that govern complex traits is pivotal for advancing crop improvement. Here we present a comprehensive regulome atlas for rice (Oryza sativa), charting the chromatin accessibility across 23 distinct tissues from three representative varieties. Our study uncovers 117,176 unique open chromatin regions (OCRs), accounting for ~15% of the rice genome, a notably higher proportion compared to previous reports in plants. Integrating RNA-seq data from matched tissues, we confidently predict 59,075 OCR-to-gene links, with enhancers constituting 69.54% of these associations, including many known enhancer-to-gene links. Leveraging this resource, we re-evaluate genome-wide association study results and discover a previously unknown function of OsbZIP06 in seed germination, which we subsequently confirm through experimental validation. We optimize deep learning models to decode regulatory grammar, achieving robust modeling of tissue-specific chromatin accessibility. This approach allows to predict cross-variety regulatory dynamics from genomic sequences, shedding light on the genetic underpinnings of cis-regulatory divergence and morphological disparities between varieties. Overall, our study establishes a foundational resource for rice functional genomics and precision molecular breeding, providing valuable insights into regulatory mechanisms governing complex traits.
doi: 10.1038/s41467-024-50787-y PubMed: 39095348 Google Scholar

72. cisDynet: An integrated platform for modeling gene-regulatory dynamics and networks.
Zhu T, Zhou X, You Y, Wang L, He Z, Chen D*
Imeta. 2023 Nov 23;2(4):e152. doi: 10.1002/imt2.152. eCollection 2023 Nov.
Chromatin accessibility sequencing has been widely used for uncovering genetic regulatory mechanisms and inferring gene regulatory networks. However, effectively integrating large-scale chromatin accessibility datasets has posed a significant challenge. This is due to the lack of a comprehensive end-to-end solution, as many existing tools primarily emphasize data preprocessing and overlook downstream analyses. To bridge this gap, we have introduced cisDynet, a holistic solution that combines streamlined data preprocessing using Snakemake and R functions with advanced downstream analysis capabilities. cisDynet excels in conventional data analyses, encompassing peak statistics, peak annotation, differential analysis, motif enrichment analysis, and more. Additionally, it allows to perform sophisticated data exploration, such as tissue-specific peak identification, time course data modeling, integration of RNA-seq data to establish peak-to-gene associations, constructing regulatory networks, and conducting enrichment analysis of genome-wide association study (GWAS) variants. As a proof of concept, we applied cisDynet to reanalyze comprehensive ATAC-seq datasets across various tissues from the Encyclopedia of DNA Elements (ENCODE) project. The analysis successfully delineated tissue-specific open chromatin regions (OCRs), established connections between OCRs and target genes, and effectively linked these discoveries with 1861 GWAS variants. Furthermore, cisDynet was instrumental in dissecting the time course open chromatin data of mouse embryonic development, revealing the dynamic behavior of OCRs over developmental stages and identifying key transcription factors governing differentiation trajectories. In summary, cisDynet offers researchers a user-friendly solution that minimizes the need for extensive coding, ensures the reproducibility of results, and greatly simplifies the exploration of epigenomic data.

71. Comprehensive integration of single-cell transcriptomic data illuminates the regulatory network architecture of plant cell fate specification.
Cao S, Zhao X, Li Z, Yu R, Li Y, Zhou X, Yan W, Chen D*, He C
Plant Divers. 2024 Apr 3;46(3):372-385. doi: 10.1016/j.pld.2024.03.008. eCollection 2024 May.
Plant morphogenesis relies on precise gene expression programs at the proper time and position which is orchestrated by transcription factors (TFs) in intricate regulatory networks in a cell-type specific manner. Here we introduced a comprehensive single-cell transcriptomic atlas of Arabidopsis seedlings. This atlas is the result of meticulous integration of 63 previously published scRNA-seq datasets, addressing batch effects and conserving biological variance. This integration spans a broad spectrum of tissues, including both below- and above-ground parts. Utilizing a rigorous approach for cell type annotation, we identified 47 distinct cell types or states, largely expanding our current view of plant cell compositions. We systematically constructed cell-type specific gene regulatory networks and uncovered key regulators that act in a coordinated manner to control cell-type specific gene expression. Taken together, our study not only offers extensive plant cell atlas exploration that serves as a valuable resource, but also provides molecular insights into gene-regulatory programs that varies from different cell types.
doi: 10.1016/j.pld.2024.03.008 PubMed: 38798726 Google Scholar

70. Dissecting the molecular basis of spike traits by integrating gene regulatory networks and genetic variation in wheat.
Ai G, He C, Bi S, Zhou Z, Liu A, Hu X, Liu Y, Jin L, Zhou J, Zhang H, Du D, Chen H, Gong X, Saeed S, Su H, Lan C, Chen W, Li Q, Mao H, Li L, Liu H, Chen D, Kaufmann K, Alazab KF, Yan W
Plant Commun. 2024 May 13;5(5):100879. doi: 10.1016/j.xplc.2024.100879. Epub 2024 Mar 14.
Spike architecture influences both grain weight and grain number per spike, which are the two major components of grain yield in bread wheat (Triticum aestivum L.). However, the complex wheat genome and the influence of various environmental factors pose challenges in mapping the causal genes that affect spike traits. Here, we systematically identified genes involved in spike trait formation by integrating information on genomic variation and gene regulatory networks controlling young spike development in wheat. We identified 170 loci that are responsible for variations in spike length, spikelet number per spike, and grain number per spike through genome-wide association study and meta-QTL analyses. We constructed gene regulatory networks for young inflorescences at the double ridge stage and the floret primordium stage, in which the spikelet meristem and the floret meristem are predominant, respectively, by integrating transcriptome, histone modification, chromatin accessibility, eQTL, and protein-protein interactome data. From these networks, we identified 169 hub genes located in 76 of the 170 QTL regions whose polymorphisms are significantly associated with variation in spike traits. The functions of TaZF-B1, VRT-B2, and TaSPL15-A/D in establishment of wheat spike architecture were verified. This study provides valuable molecular resources for understanding spike traits and demonstrates that combining genetic analysis and developmental regulatory networks is a robust approach for dissection of complex traits.
doi: 10.1016/j.xplc.2024.100879 PubMed: 38486454 Google Scholar

69. Capture of regulatory factors via CRISPR-dCas9 for mechanistic analysis of fine-tuned SERRATE expression in Arabidopsis.
Chen W, Wang J, Wang Z, Zhu T, Zheng Y, Hawar A, Chang Y, Wang X, Li D, Wang G, Yang W, Zhao Y, Chen D, Yuan YA, Sun B
Nat Plants. 2024 Jan;10(1):86-99. doi: 10.1038/s41477-023-01575-x. Epub 2024 Jan 2.
SERRATE (SE) plays an important role in many biological processes and under biotic stress resistance. However, little about the control of SE has been clarified. Here we present a method named native chromatin-associated proteome affinity by CRISPR-dCas9 (CASPA-dCas9) to holistically capture native regulators of the SE locus. Several key regulatory factors including PHYTOCHROME RAPIDLY REGULATED 2 (PAR2), WRKY DNA-binding protein 19 (WRKY19) and the MYB-family protein MYB27 of SE are identified. MYB27 recruits the long non-coding RNA-PRC2 (SEAIR-PRC2) complex for H3K27me3 deposition on exon 1 of SE and subsequently represses SE expression, while PAR2-MYB27 interaction inhibits both the binding of MYB27 on the SE promoter and the recruitment of SEAIR-PRC2 by MYB27. The interaction between PAR2 and MYB27 fine-tunes the SE expression level at different developmental stages. In addition, PAR2 and WRKY19 synergistically promote SE expression for pathogen resistance. Collectively, our results demonstrate an efficient method to capture key regulators of target genes and uncover the precise regulatory mechanism for SE.
doi: 10.1038/s41477-023-01575-x PubMed: 38168608 Google Scholar
2023

68. A chromosome-level genome assembly provides insights into Cornus wilsoniana evolution, oil biosynthesis, and floral bud development.
He Z, Chao H, Zhou X, Ni Q, Hu Y, Yu R, Wang M, Li C, Chen J, Chen Y, Chen Y, Cui C, Zhang L, Chen M, Chen D*
Hortic Res. 2023 Sep 29;10(11):uhad196. doi: 10.1093/hr/uhad196. eCollection 2023 Nov.
Cornus wilsoniana W. is a woody oil plant with high oil content and strong hypolipidemic effects, making it a valuable species for medicinal, landscaping, and ecological purposes in China. To advance genetic research on this species, we employed PacBio together with Hi-C data to create a draft genome assembly for C. wilsoniana. Based on an 11-chromosome anchored chromosome-level assembly, the estimated genome size was determined to be 843.51 Mb. The N50 contig size and N50 scaffold size were calculated to be 4.49 and 78.00 Mb, respectively. Furthermore, 30 474 protein-coding genes were annotated. Comparative genomics analysis revealed that C. wilsoniana diverged from its closest species ~12.46 million years ago (Mya). Furthermore, the divergence between Cornaceae and Nyssaceae occurred >62.22 Mya. We also found evidence of whole-genome duplication events and whole-genome triplication γ, occurring at ~44.90 and 115.86 Mya. We further inferred the origins of chromosomes, which sheds light on the complex evolutionary history of the karyotype of C. wilsoniana. Through transcriptional and metabolic analysis, we identified two FAD2 homologous genes that may play a crucial role in controlling the oleic to linoleic acid ratio. We further investigated the correlation between metabolites and genes and identified 33 MADS-TF homologous genes that may affect flower morphology in C. wilsoniana. Overall, this study lays the groundwork for future research aimed at identifying the genetic basis of crucial traits in C. wilsoniana.

67. Single-cell transcriptome analysis dissects lncRNA-associated gene networks in Arabidopsis.
He Z, Lan Y, Zhou X, Yu B, Zhu T, Yang F, Fu LY, Chao H, Wang J, Feng RX, Zuo S, Lan W, Chen C, Chen M, Zhao X, Hu K, Chen D*
Plant Commun. 2024 Feb 12;5(2):100717. doi: 10.1016/j.xplc.2023.100717. Epub 2023 Sep 15.
The plant genome produces an extremely large collection of long noncoding RNAs (lncRNAs) that are generally expressed in a context-specific manner and have pivotal roles in regulation of diverse biological processes. Here, we mapped the transcriptional heterogeneity of lncRNAs and their associated gene regulatory networks at single-cell resolution. We generated a comprehensive cell atlas at the whole-organism level by integrative analysis of 28 published single-cell RNA sequencing (scRNA-seq) datasets from juvenile Arabidopsis seedlings. We then provided an in-depth analysis of cell-type-related lncRNA signatures that show expression patterns consistent with canonical protein-coding gene markers. We further demonstrated that the cell-type-specific expression of lncRNAs largely explains their tissue specificity. In addition, we predicted gene regulatory networks on the basis of motif enrichment and co-expression analysis of lncRNAs and mRNAs, and we identified putative transcription factors orchestrating cell-type-specific expression of lncRNAs. The analysis results are available at the single-cell-based plant lncRNA atlas database (scPLAD; https://biobigdata.nju.edu.cn/scPLAD/). Overall, this work demonstrates the power of integrative single-cell data analysis applied to plant lncRNA biology and provides fundamental insights into lncRNA expression specificity and associated gene regulation.
doi: 10.1016/j.xplc.2023.100717 PubMed: 37715446 Google Scholar
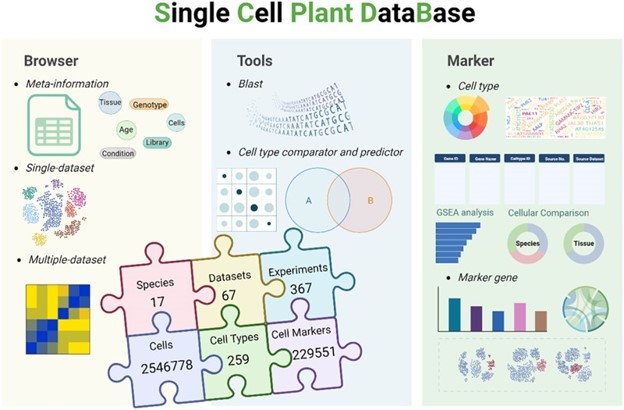
66. scPlantDB: a comprehensive database for exploring cell types and markers of plant cell atlases.
He Z, Luo Y, Zhou X, Zhu T, Lan Y, Chen D*
Nucleic Acids Res. 2024 Jan 5;52(D1):D1629-D1638. doi: 10.1093/nar/gkad706.
Recent advancements in single-cell RNA sequencing (scRNA-seq) technology have enabled the comprehensive profiling of gene expression patterns at the single-cell level, offering unprecedented insights into cellular diversity and heterogeneity within plant tissues. In this study, we present a systematic approach to construct a plant single-cell database, scPlantDB, which is publicly available at https://biobigdata.nju.edu.cn/scplantdb. We integrated single-cell transcriptomic profiles from 67 high-quality datasets across 17 plant species, comprising approximately 2.5 million cells. The data underwent rigorous collection, manual curation, strict quality control and standardized processing from public databases. scPlantDB offers interactive visualization of gene expression at the single-cell level, facilitating the exploration of both single-dataset and multiple-dataset analyses. It enables systematic comparison and functional annotation of markers across diverse cell types and species while providing tools to identify and compare cell types based on these markers. In summary, scPlantDB serves as a comprehensive database for investigating cell types and markers within plant cell atlases. It is a valuable resource for the plant research community.

65. Single cell transcriptomic analyses implicate an immunosuppressive tumor microenvironment in pancreatic cancer liver metastasis.
Zhang S#, Fang W#, Zhou S#, Zhu D#, Chen R#, Gao X, Li Z, Fu Y, Zhang Y, Yang F, Zhao J, Wu H, Wang P, Shen Y, Shen S, Xu G, Wang L, Yan C, Zou X, Chen D*, Lv Y
Nat Commun. 2023 Aug 23;14(1):5123. doi: 10.1038/s41467-023-40727-7.
Pancreatic ductal adenocarcinoma (PDAC) is a highly metastatic disease refractory to all targeted and immune therapies. However, our understanding of PDAC microenvironment especially the metastatic microenvironment is very limited partly due to the inaccessibility to metastatic tumor tissues. Here, we present the single-cell transcriptomic landscape of synchronously resected PDAC primary tumors and matched liver metastases. We perform comparative analysis on both cellular composition and functional phenotype between primary and metastatic tumors. Tumor cells exhibit distinct transcriptomic profile in liver metastasis with clearly defined evolutionary routes from cancer cells in primary tumor. We also identify specific subtypes of stromal and immune cells critical to the formation of the pro-tumor microenvironment in metastatic lesions, including RGS5+ cancer-associated fibroblasts, CCL18+ lipid-associated macrophages, S100A8+ neutrophils and FOXP3+ regulatory T cells. Cellular interactome analysis further reveals that the lack of tumor-immune cell interaction in metastatic tissues contributes to the formation of the immunosuppressive microenvironment. Our study provides a comprehensive characterization of the transcriptional landscape of PDAC liver metastasis.
doi: 10.1038/s41467-023-40727-7 PubMed: 37612267 Google Scholar
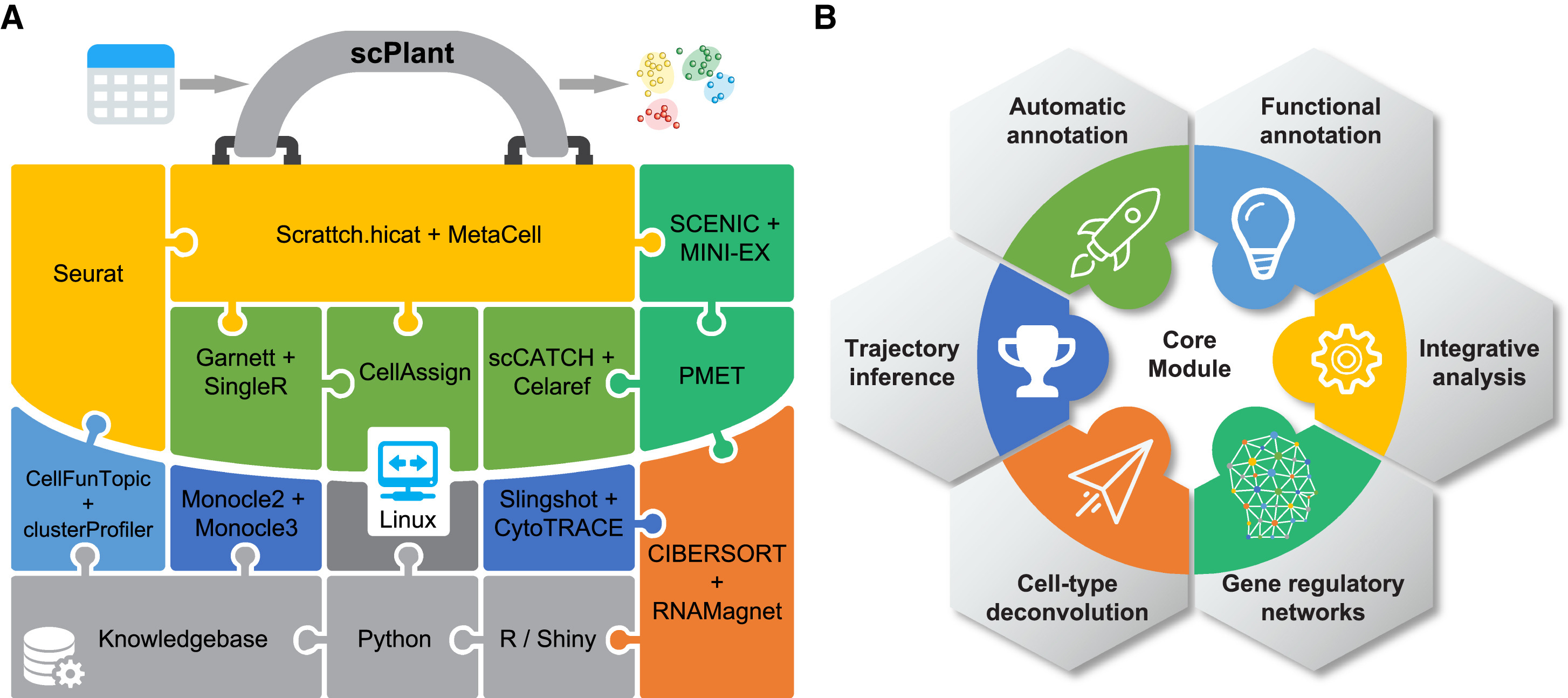
64. scPlant: A versatile framework for single-cell transcriptomic data analysis in plants.
Cao S, He Z, Chen R, Luo Y, Fu LY, Zhou X, He C, Yan W, Zhang CY, Chen D*
Plant Commun. 2023 Sep 11;4(5):100631. doi: 10.1016/j.xplc.2023.100631. Epub 2023 May 29.
Single-cell transcriptomics has been fully embraced in plant biological research and is revolutionizing our understanding of plant growth, development, and responses to external stimuli. However, single-cell transcriptomic data analysis in plants is not trivial, given that there is currently no end-to-end solution and that integration of various bioinformatics tools involves a large number of required dependencies. Here, we present scPlant, a versatile framework for exploring plant single-cell atlases with minimum input data provided by users. The scPlant pipeline is implemented with numerous functions for diverse analytical tasks, ranging from basic data processing to advanced demands such as cell-type annotation and deconvolution, trajectory inference, cross-species data integration, and cell-type-specific gene regulatory network construction. In addition, a variety of visualization tools are bundled in a built-in Shiny application, enabling exploration of single-cell transcriptomic data on the fly.
doi: 10.1016/j.xplc.2023.100631 PubMed: 37254480 Google Scholar
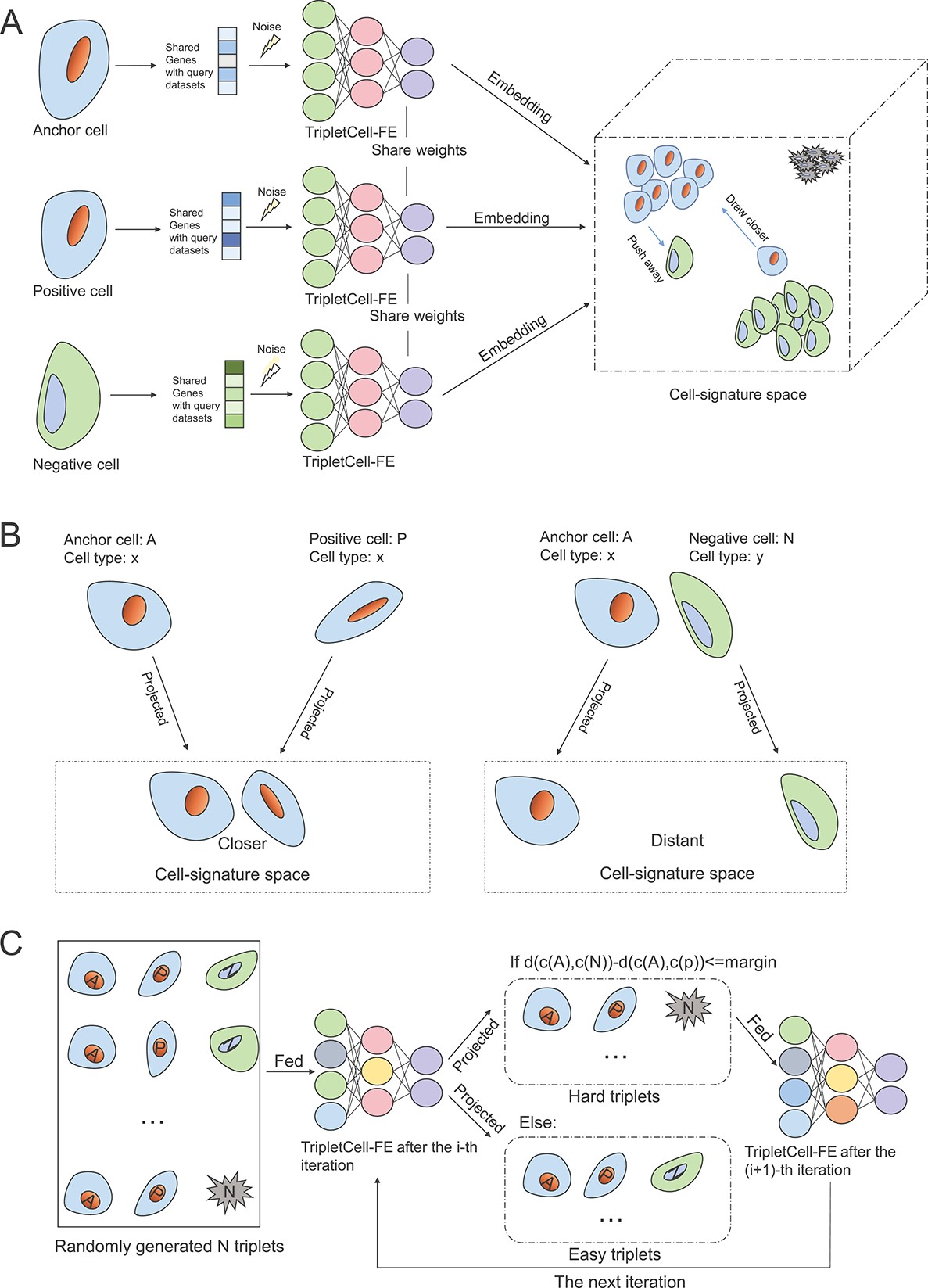
63. TripletCell: a deep metric learning framework for accurate annotation of cell types at the single-cell level.
Liu Y, Wei G, Li C, Shen LC, Gasser RB, Song J, Chen D*, Yu DJ
Brief Bioinform. 2023 May 19;24(3):bbad132. doi: 10.1093/bib/bbad132.
Single-cell RNA sequencing (scRNA-seq) has significantly accelerated the experimental characterization of distinct cell lineages and types in complex tissues and organisms. Cell-type annotation is of great importance in most of the scRNA-seq analysis pipelines. However, manual cell-type annotation heavily relies on the quality of scRNA-seq data and marker genes, and therefore can be laborious and time-consuming. Furthermore, the heterogeneity of scRNA-seq datasets poses another challenge for accurate cell-type annotation, such as the batch effect induced by different scRNA-seq protocols and samples. To overcome these limitations, here we propose a novel pipeline, termed TripletCell, for cross-species, cross-protocol and cross-sample cell-type annotation. We developed a cell embedding and dimension-reduction module for the feature extraction (FE) in TripletCell, namely TripletCell-FE, to leverage the deep metric learning-based algorithm for the relationships between the reference gene expression matrix and the query cells. Our experimental studies on 21 datasets (covering nine scRNA-seq protocols, two species and three tissues) demonstrate that TripletCell outperformed state-of-the-art approaches for cell-type annotation. More importantly, regardless of protocols or species, TripletCell can deliver outstanding and robust performance in annotating different types of cells. TripletCell is freely available at https://github.com/liuyan3056/TripletCell. We believe that TripletCell is a reliable computational tool for accurately annotating various cell types using scRNA-seq data and will be instrumental in assisting the generation of novel biological hypotheses in cell biology.
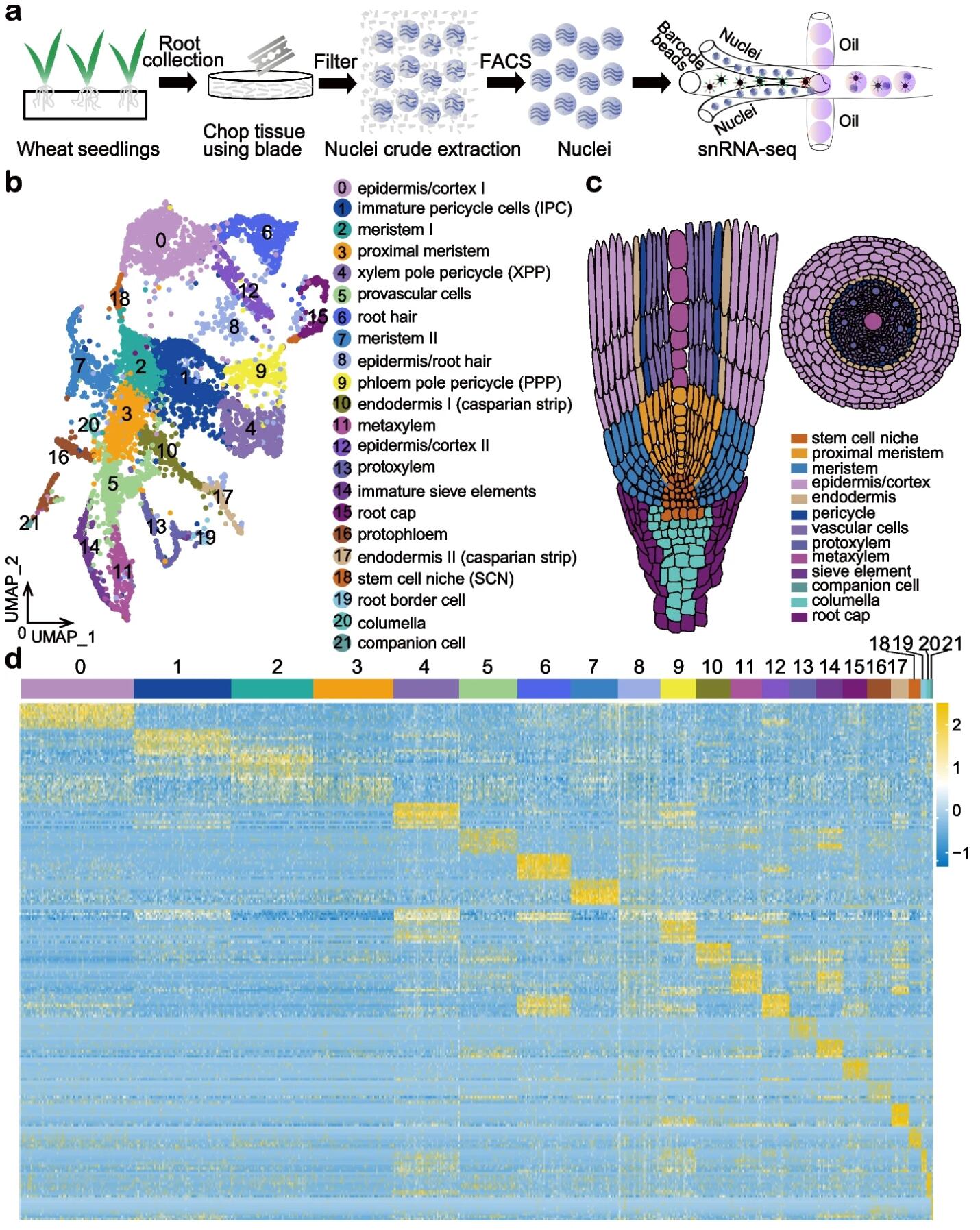
62. Asymmetric gene expression and cell-type-specific regulatory networks in the root of bread wheat revealed by single-cell multiomics analysis.
Zhang L#, He C#, Lai Y#, Wang Y, Kang L, Liu A, Lan C, Su H, Gao Y, Li Z, Yang F, Li Q, Mao H, Chen D, Chen W, Kaufmann K, Yan W
Genome Biol. 2023 Apr 4;24(1):65. doi: 10.1186/s13059-023-02908-x.
BACKGROUND: Homoeologs are defined as homologous genes resulting from allopolyploidy. Bread wheat, Triticum aestivum, is an allohexaploid species with many homoeologs. Homoeolog expression bias, referring to the relative contribution of homoeologs to the transcriptome, is critical for determining the traits that influence wheat growth and development. Asymmetric transcription of homoeologs has been so far investigated in a tissue or organ-specific manner, which could be misleading due to a mixture of cell types. RESULTS: Here, we perform single nuclei RNA sequencing and ATAC sequencing of wheat root to study the asymmetric gene transcription, reconstruct cell differentiation trajectories and cell-type-specific gene regulatory networks. We identify 22 cell types. We then reconstruct cell differentiation trajectories that suggest different origins between epidermis/cortex and endodermis, distinguishing bread wheat from Arabidopsis. We show that the ratio of asymmetrically transcribed triads varies greatly when analyzing at the single-cell level. Hub transcription factors determining cell type identity are also identified. In particular, we demonstrate that TaSPL14 participates in vasculature development by regulating the expression of BAM1. Combining single-cell transcription and chromatin accessibility data, we construct the pseudo-time regulatory network driving root hair differentiation. We find MYB3R4, REF6, HDG1, and GATAs as key regulators in this process. CONCLUSIONS: Our findings reveal the transcriptional landscape of root organization and asymmetric gene transcription at single-cell resolution in polyploid wheat.
doi: 10.1186/s13059-023-02908-x PubMed: 37016448 Google Scholar

61. Lipid-accumulated reactive astrocytes promote disease progression in epilepsy.
Chen ZP#, Wang S#, Zhao X#, Fang W#, Wang Z, Ye H, Wang MJ, Ke L, Huang T, Lv P, Jiang X, Zhang Q, Li L, Xie ST, Zhu JN, Hang C, Chen D*, Liu X, Yan C
Nat Neurosci. 2023 Apr;26(4):542-554. doi: 10.1038/s41593-023-01288-6. Epub 2023 Mar 20.
Reactive astrocytes play an important role in neurological diseases, but their molecular and functional phenotypes in epilepsy are unclear. Here, we show that in patients with temporal lobe epilepsy (TLE) and mouse models of epilepsy, excessive lipid accumulation in astrocytes leads to the formation of lipid-accumulated reactive astrocytes (LARAs), a new reactive astrocyte subtype characterized by elevated APOE expression. Genetic knockout of APOE inhibited LARA formation and seizure activities in epileptic mice. Single-nucleus RNA sequencing in TLE patients confirmed the existence of a LARA subpopulation with a distinct molecular signature. Functional studies in epilepsy mouse models and human brain slices showed that LARAs promote neuronal hyperactivity and disease progression. Targeting LARAs by intervention with lipid transport and metabolism could thus provide new therapeutic options for drug-resistant TLE.
doi: 10.1038/s41593-023-01288-6 PubMed: 36941428 Google Scholar
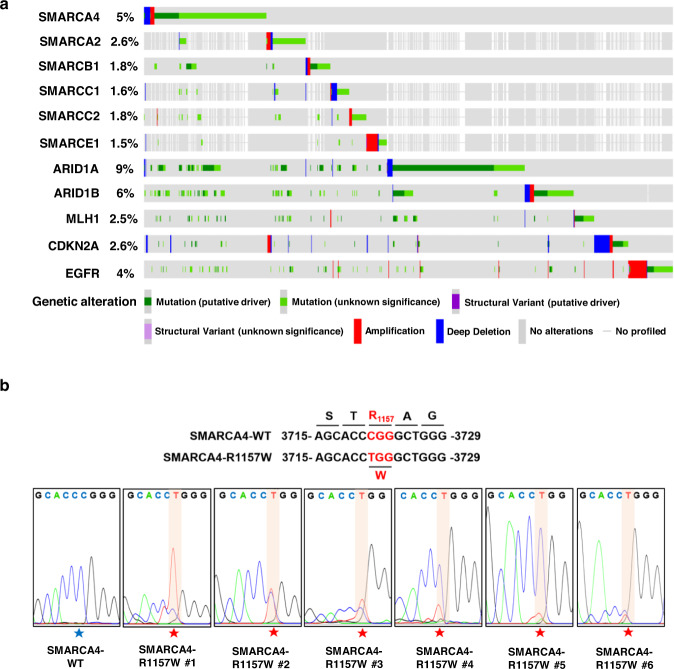
60. The SMARCA4(R1157W) mutation facilitates chromatin remodeling and confers PRMT1/SMARCA4 inhibitors sensitivity in colorectal cancer.
Zeng X#, Yao B#, Liu J#, Gong GW, Liu M, Li J, Pan HF, Li Q, Yang D, Lu P, Wu D, Xu P, Chen B, Chen P, Zhang M, Zen K, Jing J, Huang DCS, Chen D, Jiang ZW, Zhao Q
NPJ Precis Oncol. 2023 Mar 15;7(1):28. doi: 10.1038/s41698-023-00367-y.
Genomic studies have demonstrated a high frequency of genetic alterations in components of the SWI/SNF complex including the core subunit SMARCA4. However, the mechanisms of tumorigenesis driven by SMARCA4 mutations, particularly in colorectal cancer (CRC), remain largely unknown. In this study, we identified a specific, hotspot mutation in SMARCA4 (c. 3721C>T) which results in a conversion from arginine to tryptophan at residue 1157 (R1157W) in human CRC tissues associated with higher-grade tumors and controls CRC progression. Mechanistically, we found that the SMARCA4R1157W mutation facilitated its recruitment to PRMT1-mediated H4R3me2a (asymmetric dimethylation of Arg 3 in histone H4) and enhanced the ATPase activity of SWI/SNF complex to remodel chromatin in CRC cells. We further showed that the SMARCA4R1157W mutant reinforced the transcriptional expression of EGFR and TNS4 to promote the proliferation of CRC cells and patient-derived tumor organoids. Importantly, we demonstrated that SMARCA4R1157W CRC cells and mutant cell-derived xenografts were more sensitive to the combined inhibition of PRMT1 and SMARCA4 which act synergistically to suppress cell proliferation. Together, our findings show that SMARCA4-R1157W is a critical activating mutation, which accelerates CRC progression through facilitating chromatin recruitment and remodeling. Our results suggest a potential precision therapeutic strategy for the treatment of CRC patients carrying the SMARCA4R1157W mutation.
doi: 10.1038/s41698-023-00367-y PubMed: 36922568 Google Scholar
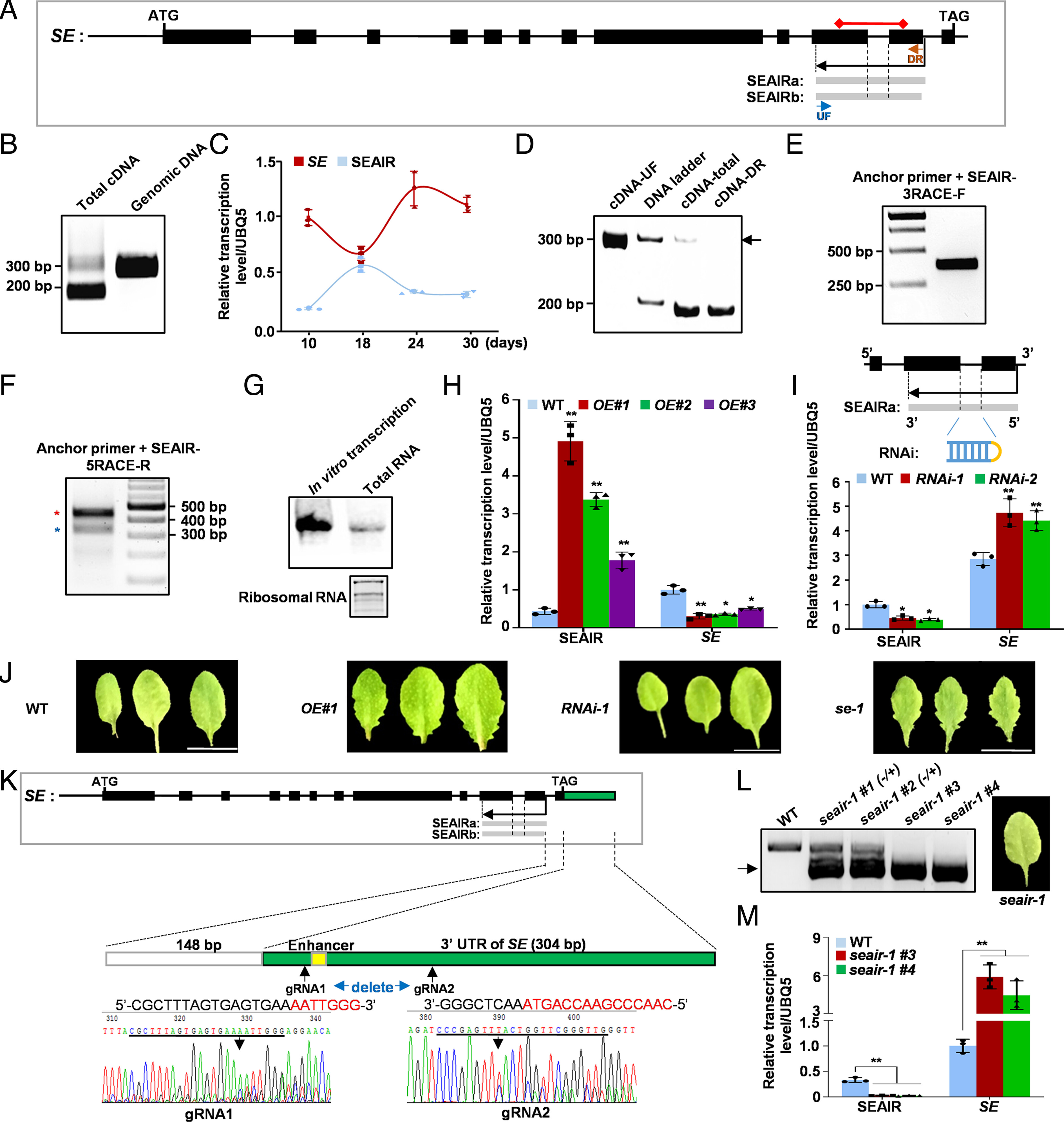
59. An antisense intragenic lncRNA SEAIRa mediates transcriptional and epigenetic repression of SERRATE in Arabidopsis.
Chen W, Zhu T, Shi Y, Chen Y, Li WJ, Chan RJ, Chen D, Zhang W, Yuan YA, Wang X, Sun B
Proc Natl Acad Sci U S A. 2023 Mar 7;120(10):e2216062120. doi: 10.1073/pnas.2216062120. Epub 2023 Mar 1.
SERRATE (SE) is a core protein for microRNA (miRNA) biogenesis as well as for mRNA alternative splicing. Investigating the regulatory mechanism of SE expression is hence critical to understanding its detailed function in diverse biological processes. However, little about the control of SE expression has been clarified, especially through long noncoding RNA (lncRNA). Here, we identified an antisense intragenic lncRNA transcribed from the 3' end of SE, named SEAIRa. SEAIRa repressed SE expression, which in turn led to serrated leaves. SEAIRa recruited plant U-box proteins PUB25/26 with unreported RNA binding ability and a ubiquitin-like protein related to ubiquitin 1 (RUB1) for H2A monoubiquitination (H2Aub) at exon 11 of SE. In addition, PUB25/26 helped cleave SEAIRa and release the 5' domain fragment, which recruited the PRC2 complex for H3 lysine 27 trimethylation (H3K27me3) deposition at the first exon of SE. The distinct modifications of H2Aub and H3K27me3 at different sites of the SE locus cooperatively suppressed SE expression. Collectively, our results uncover an epigenetic mechanism mediated by the lncRNA SEAIRa that modulates SE expression, which is indispensable for plant growth and development.
doi: 10.1073/pnas.2216062120 PubMed: 36857348 Google Scholar
2022
58. Mapping open chromatin by ATAC-seq in bread wheat.
Wang X, Chen C, He C, Chen D, Yan W
Front Plant Sci. 2022 Nov 16;13:1074873. doi: 10.3389/fpls.2022.1074873. eCollection 2022.
Gene transcription is largely regulated by cis-regulatory elements. Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) is an emerging technology that can accurately map cis-regulatory elements in animals and plants. However, the presence of cell walls and chloroplasts in plants hinders the extraction of high-quality nuclei, thereby affects the quality of ATAC-seq data. Meanwhile, it is tricky to perform ATAC-seq with different tissue types, especially for those with limited size and amount. Moreover, with rapid growth of ATAC-seq datasets from plants, powerful and easy-to-use data analysis pipelines for ATAC-seq, especially for wheat is lacking. Here, we provided an all-in-one solution for mapping open chromatin in wheat including both experimental and data analysis procedure. We efficiently obtained nuclei with less cell debris from various wheat tissues. High-quality ATAC-seq data from young spike and ovary, which are hard to harvest were generated. We determined that the saturation sequencing depth of wheat ATAC-seq is about 16 Gb. Particularly, we developed a powerful and easy-to-use online pipeline to analyze the wheat ATAC-seq data and this pipeline can be easily extended to other plant species. The method developed here will facilitate plant regulatory genome study not only for wheat but also for other plant species.
doi: 10.3389/fpls.2022.1074873 PubMed: 36466281 Google Scholar

57. Exploring long non-coding RNA networks from single cell omics data.
Zhao X, Lan Y, Chen D*
Comput Struct Biotechnol J. 2022 Aug 4;20:4381-4389. doi: 10.1016/j.csbj.2022.08.003. eCollection 2022.
Single-cell omics technologies provide an unprecedented opportunity to decipher molecular mechanisms underlying various biological processes in a cellular heterogeneity manner. The emergence of such techniques promotes the exploration of lncRNAs, which are known to be tissue- and cell-specific noncoding transcripts involving the regulation of multiple important cellular processes. In this review, we introduce the advancement of lncRNA studies which benefit from single-cell omics data analysis. We discuss the expression heterogeneity of lncRNAs, their cell-type specificity and associated gene regulatory networks (GRNs) from a single-cell perspective. We also summarized the state-of-the-art single-cell omics resources and tools for the construction of single-cell GRNs (scGRNs) that could be potentially used for lncRNA functional study. Finally, we highlight the challenges and prospective for scGRN exploration in lncRNA biology.
doi: 10.1016/j.csbj.2022.08.003 PubMed: 36051880 Google Scholar

56. A reference single-cell regulomic and transcriptomic map of cynomolgus monkeys.
Qu J#, Yang F#, Zhu T#, Wang Y#, Fang W, Ding Y, Zhao X, Qi X, Xie Q, Chen M, Xu Q, Xie Y, Sun Y, Chen D*
Nat Commun. 2022 Jul 13;13(1):4069. doi: 10.1038/s41467-022-31770-x.
Non-human primates are attractive laboratory animal models that accurately reflect both developmental and pathological features of humans. Here we present a compendium of cell types across multiple organs in cynomolgus monkeys (Macaca fascicularis) using both single-cell chromatin accessibility and RNA sequencing data. The integrated cell map enables in-depth dissection and comparison of molecular dynamics, cell-type compositions and cellular heterogeneity across multiple tissues and organs. Using single-cell transcriptomic data, we infer pseudotime cell trajectories and cell-cell communications to uncover key molecular signatures underlying their cellular processes. Furthermore, we identify various cell-specific cis-regulatory elements and construct organ-specific gene regulatory networks at the single-cell level. Finally, we perform comparative analyses of single-cell landscapes among mouse, monkey and human. We show that cynomolgus monkey has strikingly higher degree of similarities in terms of immune-associated gene expression patterns and cellular communications to human than mouse. Taken together, our study provides a valuable resource for non-human primate cell biology.
doi: 10.1038/s41467-022-31770-x PubMed: 35831300 Google Scholar

55. ChIP-Hub provides an integrative platform for exploring plant regulome.
Fu LY#, Zhu T#, Zhou X#, Yu R#, He Z, Zhang P, Wu Z, Chen M, Kaufmann K, Chen D*
Nat Commun. 2022 Jun 14;13(1):3413. doi: 10.1038/s41467-022-30770-1.
Plant genomes encode a complex and evolutionary diverse regulatory grammar that forms the basis for most life on earth. A wealth of regulome and epigenome data have been generated in various plant species, but no common, standardized resource is available so far for biologists. Here, we present ChIP-Hub, an integrative web-based platform in the ENCODE standards that bundles >10,000 publicly available datasets reanalyzed from >40 plant species, allowing visualization and meta-analysis. We manually curate the datasets through assessing ~540 original publications and comprehensively evaluate their data quality. As a proof of concept, we extensively survey the co-association of different regulators and construct a hierarchical regulatory network under a broad developmental context. Furthermore, we show how our annotation allows to investigate the dynamic activity of tissue-specific regulatory elements (promoters and enhancers) and their underlying sequence grammar. Finally, we analyze the function and conservation of tissue-specific promoters, enhancers and chromatin states using comparative genomics approaches. Taken together, the ChIP-Hub platform and the analysis results provide rich resources for deep exploration of plant ENCODE. ChIP-Hub is available at https://biobigdata.nju.edu.cn/ChIPHub/ .
doi: 10.1038/s41467-022-30770-1 PubMed: 35701419 Google Scholar
54. The genome of hibiscus hamabo reveals its adaptation to saline and waterlogged habitat.
Wang Z, Xue JY, Hu SY, Zhang F, Yu R, Chen D, Van de Peer Y, Jiang J, Song A, Ni L, Hua J, Lu Z, Yu C, Yin Y, Gu C
Hortic Res. 2022 Mar 23;9:uhac067. doi: 10.1093/hr/uhac067. eCollection 2022.
Hibiscus hamabo is a semi-mangrove species with strong tolerance to salt and waterlogging stress. However, the molecular basis and mechanisms that underlie this strong adaptability to harsh environments remain poorly understood. Here, we assembled a high-quality, chromosome-level genome of this semi-mangrove plant and analyzed its transcriptome under different stress treatments to reveal regulatory responses and mechanisms. Our analyses suggested that H. hamabo has undergone two recent successive polyploidy events, a whole-genome duplication followed by a whole-genome triplication, resulting in an unusually large gene number (107 309 genes). Comparison of the H. hamabo genome with that of its close relative Hibiscus cannabinus, which has not experienced a recent WGT, indicated that genes associated with high stress resistance have been preferentially preserved in the H. hamabo genome, suggesting an underlying association between polyploidy and stronger stress resistance. Transcriptomic data indicated that genes in the roots and leaves responded differently to stress. In roots, genes that regulate ion channels involved in biosynthetic and metabolic processes responded quickly to adjust the ion concentration and provide metabolic products to protect root cells, whereas no such rapid response was observed from genes in leaves. Using co-expression networks, potential stress resistance genes were identified for use in future functional investigations. The genome sequence, along with several transcriptome datasets, provide insights into genome evolution and the mechanism of salt and waterlogging tolerance in H. hamabo, suggesting the importance of polyploidization for environmental adaptation.
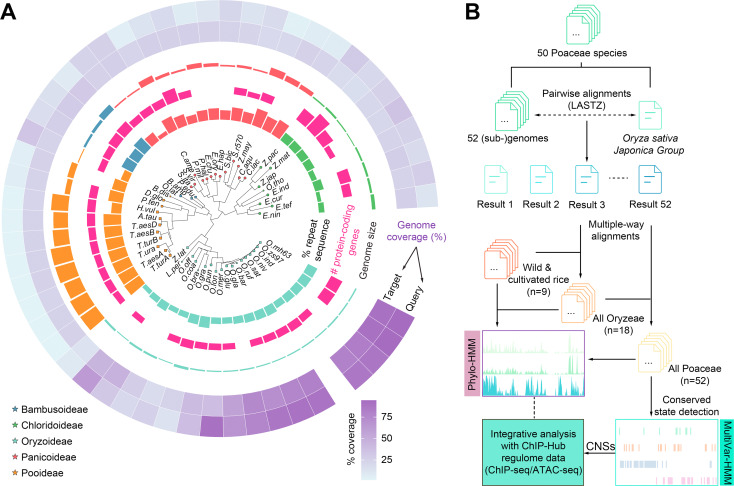
53. Systematic annotation of conservation states provides insights into regulatory regions in rice.
Zhou X, Zhu T, Fang W, Yu R, He Z, Chen D*
J Genet Genomics. 2022 Dec;49(12):1127-1137. doi: 10.1016/j.jgg.2022.04.003. Epub 2022 Apr 22.
Plant genomes contain a large fraction of noncoding sequences. The discovery and annotation of conserved noncoding sequences (CNSs) in plants is an ongoing challenge. Here we report the application of comparative genomics to systematically identify CNSs in 50 well-annotated Gramineae genomes using rice (Oryza sativa) as the reference. We conduct multiple-way whole-genome alignments to the rice genome. The rice genome is annotated as 20 conservation states (CSs) at single-nucleotide resolution using a multivariate hidden Markov model (ConsHMM) based on the multiple-genome alignments. Different states show distinct enrichments for various genomic features, and the conservation scores of CSs are highly correlated with the level of associated chromatin accessibility. We find that at least 33.5% of the rice genome is highly under selection, with more than 70% of the sequence lying outside of coding regions. A catalog of 855,366 regulatory CNSs is generated, and they significantly overlapped with putative active regulatory elements such as promoters, enhancers, and transcription factor binding sites. Collectively, our study provides a resource for elucidating functional noncoding regions of the rice genome and an evolutionary aspect of regulatory sequences in higher plants.
doi: 10.1016/j.jgg.2022.04.003 PubMed: 35470092 Google Scholar
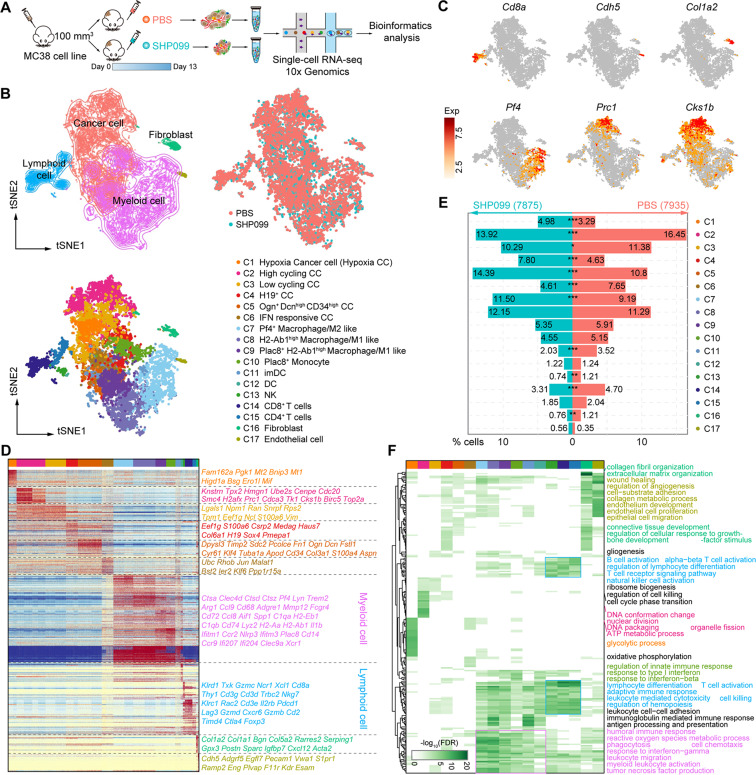
52. Allosteric inhibition reveals SHP2-mediated tumor immunosuppression in colon cancer by single-cell transcriptomics.
Gao J, Wu Z, Zhao M, Zhang R, Li M, Sun D, Cheng H, Qi X, Shen Y, Xu Q, Chen H, Chen D*, Sun Y
Acta Pharm Sin B. 2022 Jan;12(1):149-166. doi: 10.1016/j.apsb.2021.08.006. Epub 2021 Aug 11.
Colorectal cancer (CRC), a malignant tumor worldwide consists of microsatellite instability (MSI) and stable (MSS) phenotypes. Although SHP2 is a hopeful target for cancer therapy, its relationship with innate immunosuppression remains elusive. To address that, single-cell RNA sequencing was performed to explore the role of SHP2 in all cell types of tumor microenvironment (TME) from murine MC38 xenografts. Intratumoral cells were found to be functionally heterogeneous and responded significantly to SHP099, a SHP2 allosteric inhibitor. The malignant evolution of tumor cells was remarkably arrested by SHP099. Mechanistically, STING-TBK1-IRF3-mediated type I interferon signaling was highly activated by SHP099 in infiltrated myeloid cells. Notably, CRC patients with MSS phenotype exhibited greater macrophage infiltration and more potent SHP2 phosphorylation in CD68+ macrophages than MSI-high phenotypes, suggesting the potential role of macrophagic SHP2 in TME. Collectively, our data reveals a mechanism of innate immunosuppression mediated by SHP2, suggesting that SHP2 is a promising target for colon cancer immunotherapy.
doi: 10.1016/j.apsb.2021.08.006 PubMed: 35127377 Google Scholar
2021
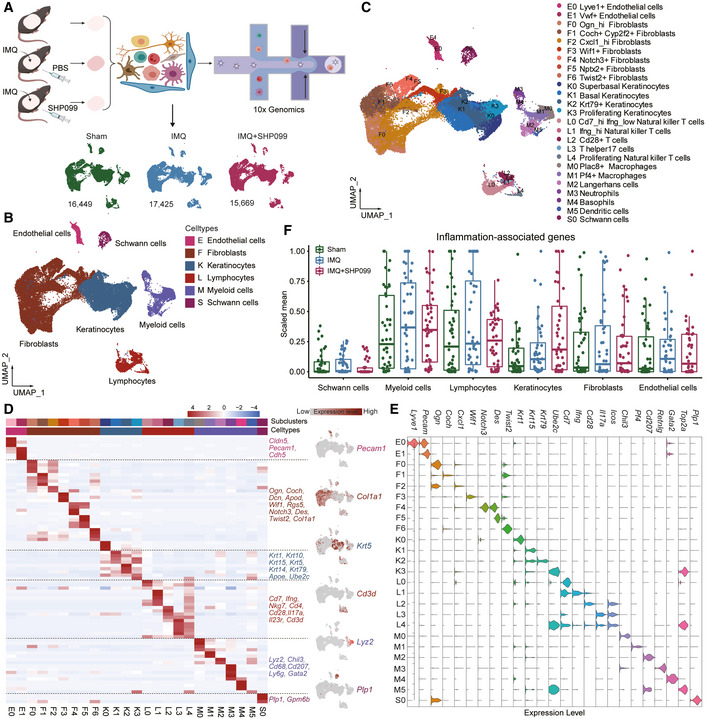
51. Allosteric inhibition of SHP2 uncovers aberrant TLR7 trafficking in aggravating psoriasis.
Zhu Y#, Wu Z#, Yan W#, Shao F#, Ke B, Jiang X, Gao J, Guo W, Lai Y, Ma H, Chen D, Xu Q, Sun Y
EMBO Mol Med. 2022 Mar 7;14(3):e14455. doi: 10.15252/emmm.202114455. Epub 2021 Dec 22.
Psoriasis is a complex chronic inflammatory skin disease with unclear molecular mechanisms. We found that the Src homology-2 domain-containing protein tyrosine phosphatase-2 (SHP2) was highly expressed in both psoriatic patients and imiquimod (IMQ)-induced psoriasis-like mice. Also, the SHP2 allosteric inhibitor SHP099 reduced pro-inflammatory cytokine expression in PBMCs taken from psoriatic patients. Consistently, SHP099 significantly ameliorated IMQ-triggered skin inflammation in mice. Single-cell RNA sequencing of murine skin demonstrated that SHP2 inhibition impaired skin inflammation in myeloid cells, especially macrophages. Furthermore, IMQ-induced psoriasis-like skin inflammation was significantly alleviated in myeloid cells (monocytes, mature macrophages, and granulocytes)-but not dendritic cells conditional SHP2 knockout mice. Mechanistically, SHP2 promoted the trafficking of toll-like receptor 7 (TLR7) from the Golgi to the endosome in macrophages by dephosphorylating TLR7 at Tyr1024, boosting the ubiquitination of TLR7 and NF-κB-mediated skin inflammation. Importantly, Tlr7 point-mutant knock-in mice showed an attenuated psoriasis-like phenotype compared to wild-type littermates following IMQ treatment. Collectively, our findings identify SHP2 as a novel regulator of psoriasis and suggest that SHP2 inhibition may be a promising therapeutic approach for psoriatic patients.
doi: 10.15252/emmm.202114455 PubMed: 34936223 Google Scholar
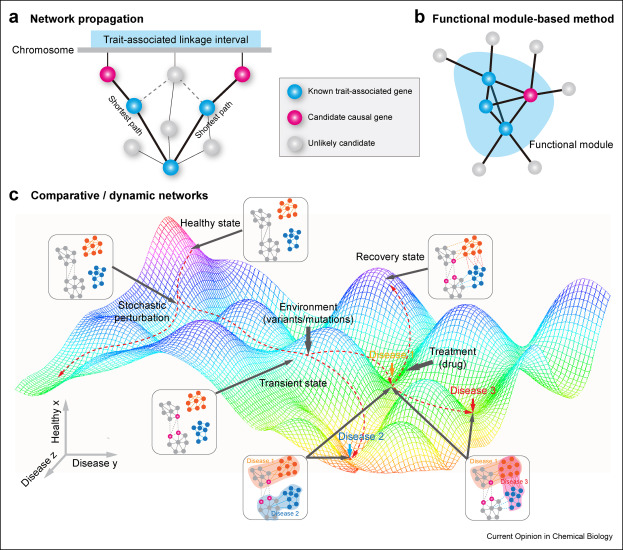
50. Network biology bridges the gaps between quantitative genetics and multi-omics to map complex diseases.
Wu S, Chen D, Snyder MP
Curr Opin Chem Biol. 2022 Feb;66:102101. doi: 10.1016/j.cbpa.2021.102101. Epub 2021 Nov 30.
With advances in high-throughput sequencing technologies, quantitative genetics approaches have provided insights into genetic basis of many complex diseases. Emerging in-depth multi-omics profiling technologies have created exciting opportunities for systematically investigating intricate interaction networks with different layers of biological molecules underlying disease etiology. Herein, we summarized two main categories of biological networks: evidence-based and statistically inferred. These different types of molecular networks complement each other at both bulk and single-cell levels. We also review three main strategies to incorporate quantitative genetics results with multi-omics data by network analysis: (a) network propagation, (b) functional module-based methods, (c) comparative/dynamic networks. These strategies not only aid in elucidating molecular mechanisms of complex diseases but can guide the search for therapeutic targets.
doi: 10.1016/j.cbpa.2021.102101 PubMed: 34861483 Google Scholar
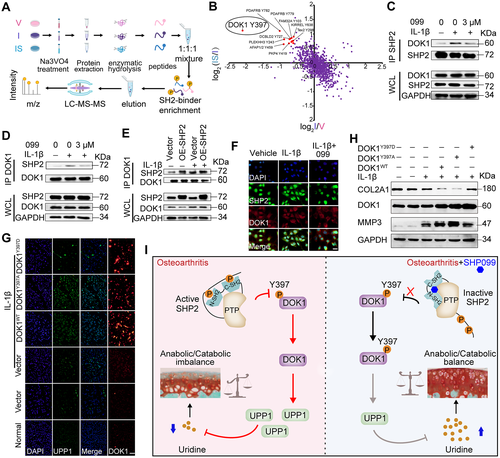
49. SH2 Domain-Containing Phosphatase 2 Inhibition Attenuates Osteoarthritis by Maintaining Homeostasis of Cartilage Metabolism via the Docking Protein 1/Uridine Phosphorylase 1/Uridine Cascade.
Liu Q, Zhai L, Han M, Shi D, Sun Z, Peng S, Wang M, Zhang C, Gao J, Yan W, Jiang Q, Chen D, Xu Q, Tan M, Sun Y
Arthritis Rheumatol. 2022 Mar;74(3):462-474. doi: 10.1002/art.41988. Epub 2022 Feb 2.
OBJECTIVE: Protein tyrosine kinases regulate osteoarthritis (OA) progression by activating a series of signal transduction pathways. However, the roles of protein tyrosine phosphatases (PTPs) in OA remain obscure. This study was undertaken to identify specific PTPs involved in OA and investigate their underlying mechanisms. METHODS: The expression of 107 PTP genes in human OA cartilage was analyzed based on a single-cell sequencing data set. The enzyme activity of the PTP SH2 domain-containing phosphatase 2 (SHP-2) was detected in primary chondrocytes after interleukin-1β (IL-1β) treatment and in human OA cartilage. Mice subjected to destabilization of the medial meniscus (DMM) and IL-1β-stimulated mouse primary chondrocytes were treated with an SHP-2 inhibitor or celecoxib (a drug used for the clinical treatment of OA). The function of SHP-2 in OA pathogenesis was further verified in Aggrecan-CreERT ;SHP2flox/flox mice. The downstream protein expression profile and dephosphorylated substrate of SHP-2 were examined by tandem mass tag labeling-based global proteomic analysis and stable isotope labeling with amino acids in cell culture-labeled tyrosine phosphoproteomic analysis, respectively. RESULTS: SHP-2 enzyme activity significantly increased in human OA samples with serious articular cartilage injury and in IL-1β-stimulated mouse chondrocytes. Pharmacologic inhibition or genetic deletion of SHP-2 ameliorated OA progression. SHP-2 inhibitors dramatically reduced the expression of cartilage degradation-related genes and simultaneously promoted the expression of cartilage synthesis-related genes. Mechanistically, SHP-2 inhibition suppressed the dephosphorylation of docking protein 1 and subsequently reduced the expression of uridine phosphorylase 1 and increased the uridine level, thereby contributing to the homeostasis of cartilage metabolism. CONCLUSION: SHP-2 is a novel accelerator of the imbalance in cartilage homeostasis. Specific inhibition of SHP-2 may ameliorate OA by maintaining the anabolic-catabolic balance.
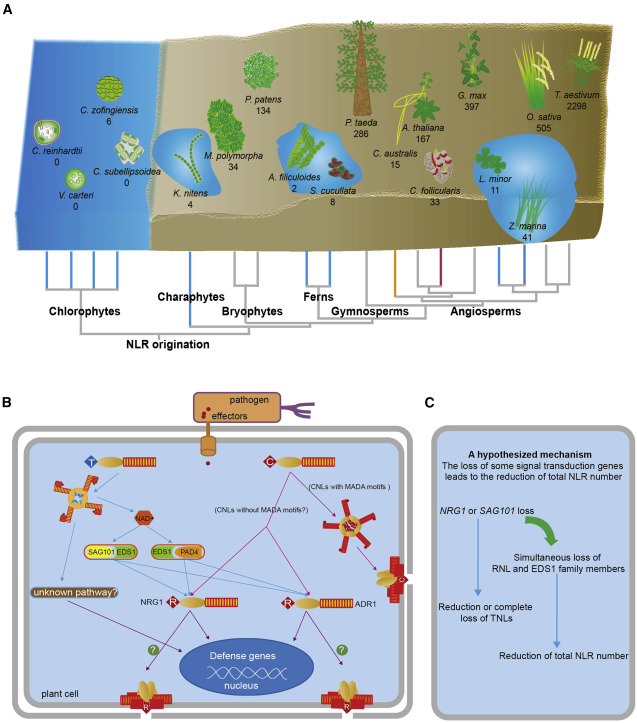
48. An angiosperm NLR Atlas reveals that NLR gene reduction is associated with ecological specialization and signal transduction component deletion.
Liu Y, Zeng Z, Zhang YM, Li Q, Jiang XM, Jiang Z, Tang JH, Chen D, Wang Q, Chen JQ, Shao ZQ
Mol Plant. 2021 Dec 6;14(12):2015-2031. doi: 10.1016/j.molp.2021.08.001. Epub 2021 Aug 4.
Nucleotide-binding leucine-rich-repeat (NLR) genes comprise the largest family of plant disease-resistance genes. Angiosperm NLR genes are phylogenetically divided into the TNL, CNL, and RNL subclasses. NLR copy numbers and subclass composition vary tremendously across angiosperm genomes. However, the evolutionary associations between genomic NLR content and ecological adaptation, or between NLR content and signal transduction components, are poorly characterized because of limited genome availability. In this study, we established an angiosperm NLR atlas (ANNA, https://biobigdata.nju.edu.cn/ANNA/) that includes NLR genes from over 300 angiosperm genomes. Using ANNA, we revealed that NLR copy numbers differ up to 66-fold among closely related species owing to rapid gene loss and gain. Interestingly, NLR contraction was associated with adaptations to aquatic, parasitic, and carnivorous lifestyles. The convergent NLR reduction in aquatic plants resembles the lack of NLR expansion during the long-term evolution of green algae before the colonization of land. A co-evolutionary pattern between NLR subclasses and plant immune pathway components was also identified, suggesting that immune pathway deficiencies may drive TNL loss. Finally, we identified a conserved TNL lineage that may function independently of the EDS1-SAG101-NRG1 module. Collectively, these findings provide new insights into the evolution of NLR genes in the context of ecological adaptation and genome content variation.
doi: 10.1016/j.molp.2021.08.001 PubMed: 34364002 Google Scholar
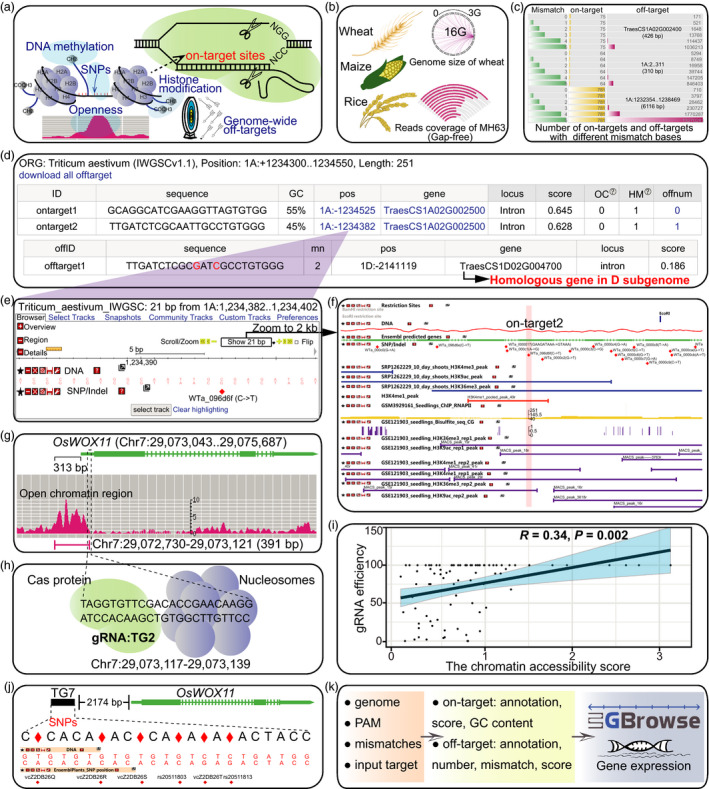
47. CRISPR-Cereal: a guide RNA design tool integrating regulome and genomic variation for wheat, maize and rice.
He C, Liu H, Chen D, Xie WZ, Wang M, Li Y, Gong X, Yan W, Chen LL
Plant Biotechnol J. 2021 Nov;19(11):2141-2143. doi: 10.1111/pbi.13675. Epub 2021 Aug 14.
2020
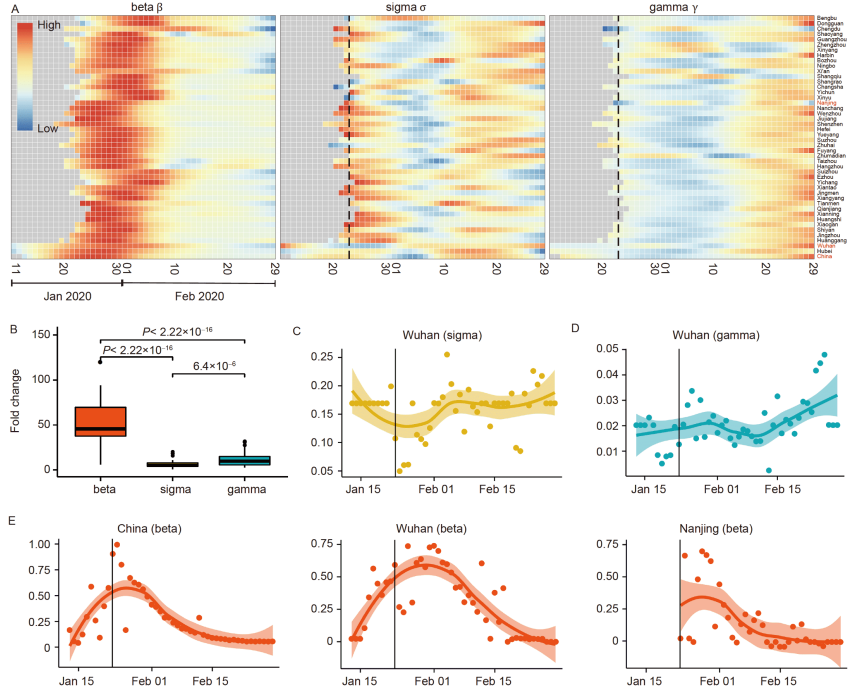
46. Modelling-based evaluation of the effect of quarantine control by the Chinese government in the coronavirus disease 2019 outbreak.
Zhou X, Wu Z, Yu R, Cao S, Fang W, Jiang Z, Yuan F, Yan C, Chen D*
Sci China Life Sci. 2020 Aug;63(8):1257-1260. doi: 10.1007/s11427-020-1717-9. Epub 2020 May 8.
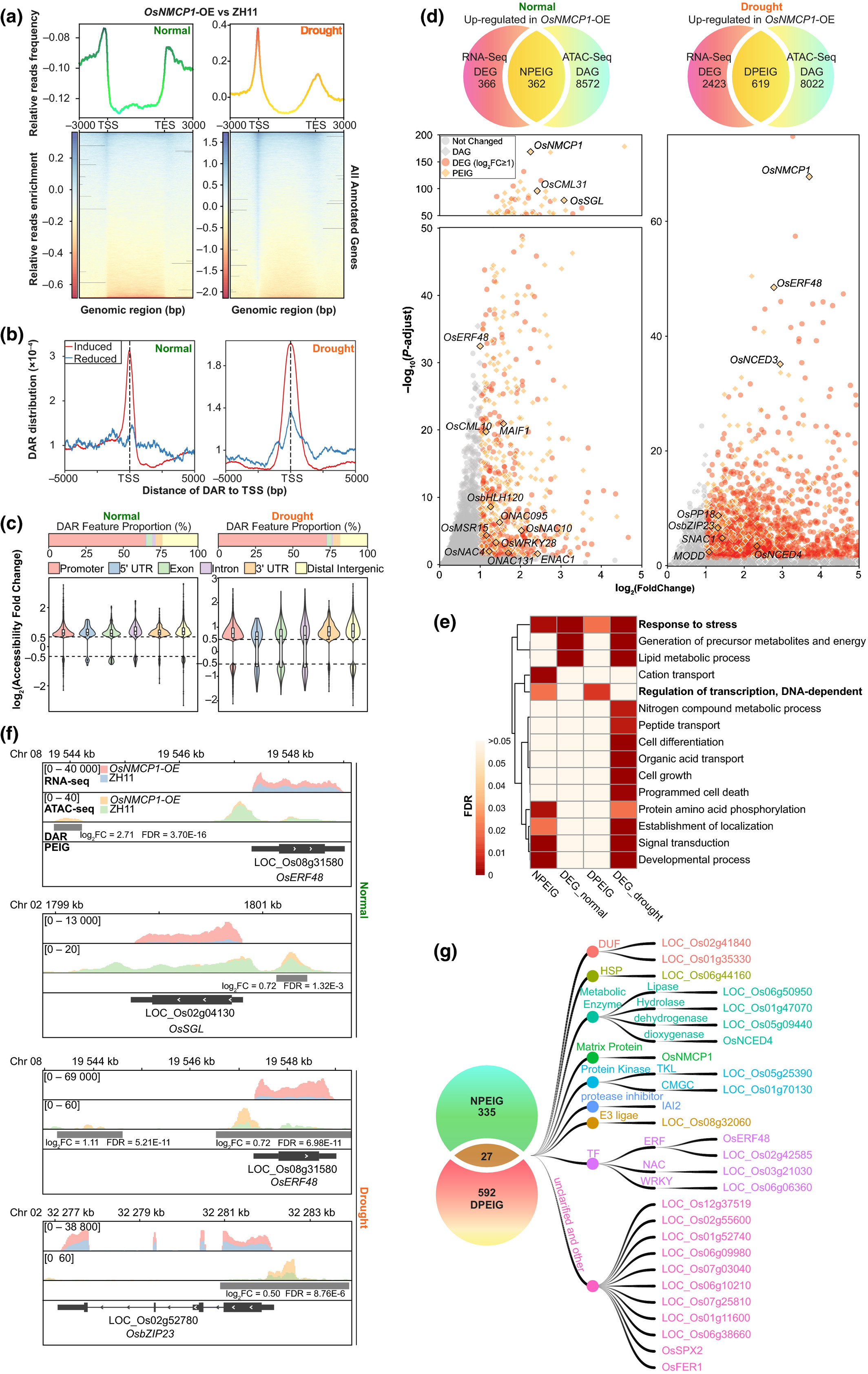
45. A lamin-like protein OsNMCP1 regulates drought resistance and root growth through chromatin accessibility modulation by interacting with a chromatin remodeller OsSWI3C in rice.
Yang J, Chang Y, Qin Y, Chen D, Zhu T, Peng K, Wang H, Tang N, Li X, Wang Y, Liu Y, Li X, Xie W, Xiong L
New Phytol. 2020 Jul;227(1):65-83. doi: 10.1111/nph.16518. Epub 2020 Apr 13.
Lamin proteins in animals are implicated in important nuclear functions, including chromatin organization, signalling transduction, gene regulation and cell differentiation. Nuclear Matrix Constituent Proteins (NMCPs) are lamin analogues in plants, but their regulatory functions remain largely unknown. We report that OsNMCP1 is localized at the nuclear periphery in rice (Oryza sativa) and induced by drought stress. OsNMCP1 overexpression resulted in a deeper and thicker root system, and enhanced drought resistance compared to the wild-type control. An assay for transposase accessible chromatin with sequencing (ATAC-seq) analysis revealed that OsNMCP1-overexpression altered chromatin accessibility in hundreds of genes related to drought resistance and root growth, including OsNAC10, OsERF48, OsSGL, SNAC1 and OsbZIP23. OsNMCP1 can interact with SWITCH/SUCROSE NONFERMENTING (SWI/SNF) chromatin remodelling complex subunit OsSWI3C. The reported drought resistance or root growth-related genes that were positively regulated by OsNMCP1 were negatively regulated by OsSWI3C under drought stress conditions, and OsSWI3C overexpression led to decreased drought resistance. We propose that the interaction between OsNMCP1 and OsSWI3C under drought stress conditions may lead to the release of OsSWI3C from the SWI/SNF gene silencing complex, thus changing chromatin accessibility in the genes related to root growth and drought resistance.
2019
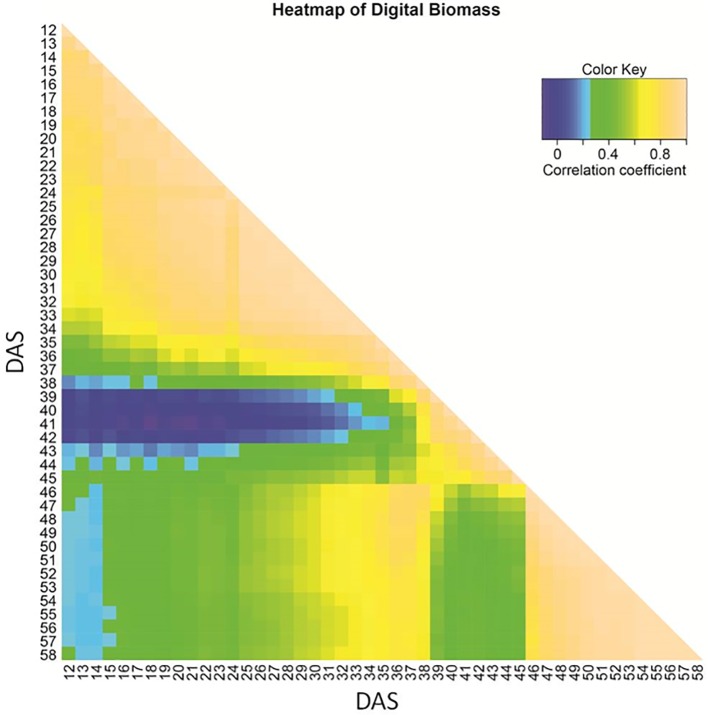
44. Non-Invasive Phenotyping Reveals Genomic Regions Involved in Pre-Anthesis Drought Tolerance and Recovery in Spring Barley.
Dhanagond S, Liu G, Zhao Y, Chen D, Grieco M, Reif J, Kilian B, Graner A, Neumann K
Front Plant Sci. 2019 Oct 25;10:1307. doi: 10.3389/fpls.2019.01307. eCollection 2019.
With ongoing climate change, drought events are becoming more frequent and will affect biomass formation when occurring during pre-flowering stages. We explored growth over time under such a drought scenario, via non-invasive imaging and revealed the underlying key genetic factors in spring barley. By comparing with well-watered conditions investigated in an earlier study and including information on timing, QTL could be classified as constitutive, drought or recovery-adaptive. Drought-adaptive QTL were found in the vicinity of genes involved in dehydration tolerance such as dehydrins (Dhn4, Dhn7, Dhn8, and Dhn9) and aquaporins (e.g. HvPIP1;5, HvPIP2;7, and HvTIP2;1). The influence of phenology on biomass formation increased under drought. Accordingly, the main QTL during recovery was the region of HvPPD-H1. The most important constitutive QTL for late biomass was located in the vicinity of HvDIM, while the main locus for seedling biomass was the HvWAXY region. The disappearance of QTL marked the genetic architecture of tiller number. The most important constitutive QTL was located on 6HS in the region of 1-FEH. Stage and tolerance specific QTL might provide opportunities for genetic manipulation to stabilize biomass and tiller number under drought conditions and thereby also grain yield.
doi: 10.3389/fpls.2019.01307 PubMed: 31708943 Google Scholar
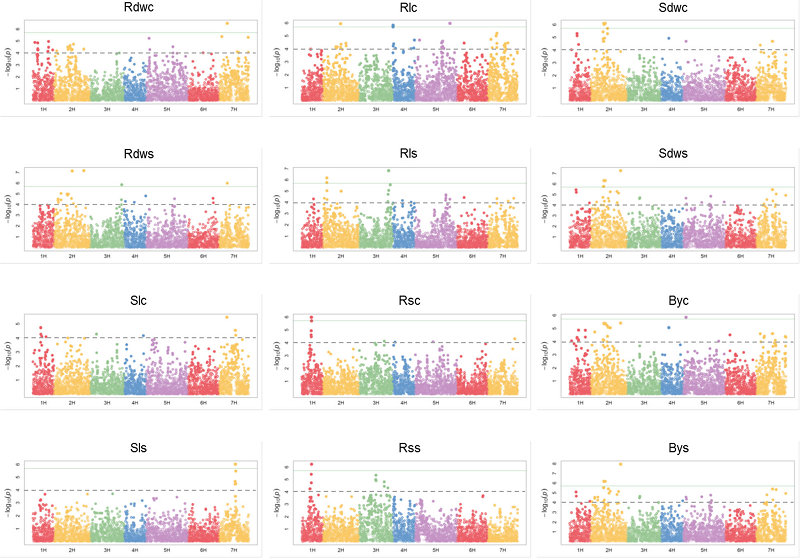
43. Genome-wide association mapping in a diverse spring barley collection reveals the presence of QTL hotspots and candidate genes for root and shoot architecture traits at seedling stage.
Abdel-Ghani AH, Sharma R, Wabila C, Dhanagond S, Owais SJ, Duwayri MA, Al-Dalain SA, Klukas C, Chen D, Lübberstedt T, von Wirén N, Graner A, Kilian B, Neumann K
BMC Plant Biol. 2019 May 23;19(1):216. doi: 10.1186/s12870-019-1828-5.
BACKGROUND: Adaptation to drought-prone environments requires robust root architecture. Genotypes with a more vigorous root system have the potential to better adapt to soils with limited moisture content. However, root architecture is complex at both, phenotypic and genetic level. Customized mapping panels in combination with efficient screenings methods can resolve the underlying genetic factors of root traits. RESULTS: A mapping panel of 233 spring barley genotypes was evaluated for root and shoot architecture traits under non-stress and osmotic stress. A genome-wide association study elucidated 65 involved genomic regions. Among them were 34 root-specific loci, eleven hotspots with associations to up to eight traits and twelve stress-specific loci. A list of candidate genes was established based on educated guess. Selected genes were tested for associated polymorphisms. By this, 14 genes were identified as promising candidates, ten remained suggestive and 15 were rejected. The data support the important role of flowering time genes, including HvPpd-H1, HvCry2, HvCO4 and HvPRR73. Moreover, seven root-related genes, HERK2, HvARF04, HvEXPB1, PIN5, PIN7, PME5 and WOX5 are confirmed as promising candidates. For the QTL with the highest allelic effect for root thickness and plant biomass a homologue of the Arabidopsis Trx-m3 was revealed as the most promising candidate. CONCLUSIONS: This study provides a catalogue of hotspots for seedling growth, root and stress-specific genomic regions along with candidate genes for future potential incorporation in breeding attempts for enhanced yield potential, particularly in drought-prone environments. Root architecture is under polygenic control. The co-localization of well-known major genes for barley development and flowering time with QTL hotspots highlights their importance for seedling growth. Association analysis revealed the involvement of HvPpd-H1 in the development of the root system. The co-localization of root QTL with HERK2, HvARF04, HvEXPB1, PIN5, PIN7, PME5 and WOX5 represents a starting point to explore the roles of these genes in barley. Accordingly, the genes HvHOX2, HsfA2b, HvHAK2, and Dhn9, known to be involved in abiotic stress response, were located within stress-specific QTL regions and await future validation.
doi: 10.1186/s12870-019-1828-5 PubMed: 31122195 Google Scholar

42. Dynamic control of enhancer activity drives stage-specific gene expression during flower morphogenesis.
Yan W, Chen D#,*, Schumacher J, Durantini D, Engelhorn J, Chen M, Carles CC, Kaufmann K
Nat Commun. 2019 Apr 12;10(1):1705. doi: 10.1038/s41467-019-09513-2.
Enhancers are critical for developmental stage-specific gene expression, but their dynamic regulation in plants remains poorly understood. Here we compare genome-wide localization of H3K27ac, chromatin accessibility and transcriptomic changes during flower development in Arabidopsis. H3K27ac prevalently marks promoter-proximal regions, suggesting that H3K27ac is not a hallmark for enhancers in Arabidopsis. We provide computational and experimental evidence to confirm that distal DNase І hypersensitive sites are predictive of enhancers. The predicted enhancers are highly stage-specific across flower development, significantly associated with SNPs for flowering-related phenotypes, and conserved across crucifer species. Through the integration of genome-wide transcription factor (TF) binding datasets, we find that floral master regulators and stage-specific TFs are largely enriched at developmentally dynamic enhancers. Finally, we show that enhancer clusters and intronic enhancers significantly associate with stage-specific gene regulation by floral master TFs. Our study provides insights into the functional flexibility of enhancers during plant development, as well as hints to annotate plant enhancers.
doi: 10.1038/s41467-019-09513-2 PubMed: 30979870 Google Scholar
2018

41. Architecture of gene regulatory networks controlling flower development in Arabidopsis thaliana.
Chen D*, Yan W, Fu LY, Kaufmann K
Nat Commun. 2018 Oct 31;9(1):4534. doi: 10.1038/s41467-018-06772-3.
Floral homeotic transcription factors (TFs) act in a combinatorial manner to specify the organ identities in the flower. However, the architecture and the function of the gene regulatory network (GRN) controlling floral organ specification is still poorly understood. In particular, the interconnections of homeotic TFs, microRNAs (miRNAs) and other factors controlling organ initiation and growth have not been studied systematically so far. Here, using a combination of genome-wide TF binding, mRNA and miRNA expression data, we reconstruct the dynamic GRN controlling floral meristem development and organ differentiation. We identify prevalent feed-forward loops (FFLs) mediated by floral homeotic TFs and miRNAs that regulate common targets. Experimental validation of a coherent FFL shows that petal size is controlled by the SEPALLATA3-regulated miR319/TCP4 module. We further show that combinatorial DNA-binding of homeotic factors and selected other TFs is predictive of organ-specific patterns of gene expression. Our results provide a valuable resource for studying molecular regulatory processes underlying floral organ specification in plants.
doi: 10.1038/s41467-018-06772-3 PubMed: 30382087 Google Scholar
40. The HTPmod Shiny application enables modeling and visualization of large-scale biological data.
Chen D*, Fu LY, Hu D, Klukas C, Chen M, Kaufmann K
Commun Biol. 2018 Jul 5;1:89. doi: 10.1038/s42003-018-0091-x.
The wave of high-throughput technologies in genomics and phenomics are enabling data to be generated on an unprecedented scale and at a reasonable cost. Exploring the large-scale data sets generated by these technologies to derive biological insights requires efficient bioinformatic tools. Here we introduce an interactive, open-source web application (HTPmod) for high-throughput biological data modeling and visualization. HTPmod is implemented with the Shiny framework by integrating the computational power and professional visualization of R and including various machine-learning approaches. We demonstrate that HTPmod can be used for modeling and visualizing large-scale, high-dimensional data sets (such as multiple omics data) under a broad context. By reinvestigating example data sets from recent studies, we find not only that HTPmod can reproduce results from the original studies in a straightforward fashion and within a reasonable time, but also that novel insights may be gained from fast reinvestigation of existing data by HTPmod.
doi: 10.1038/s42003-018-0091-x PubMed: 30271970 Google Scholar
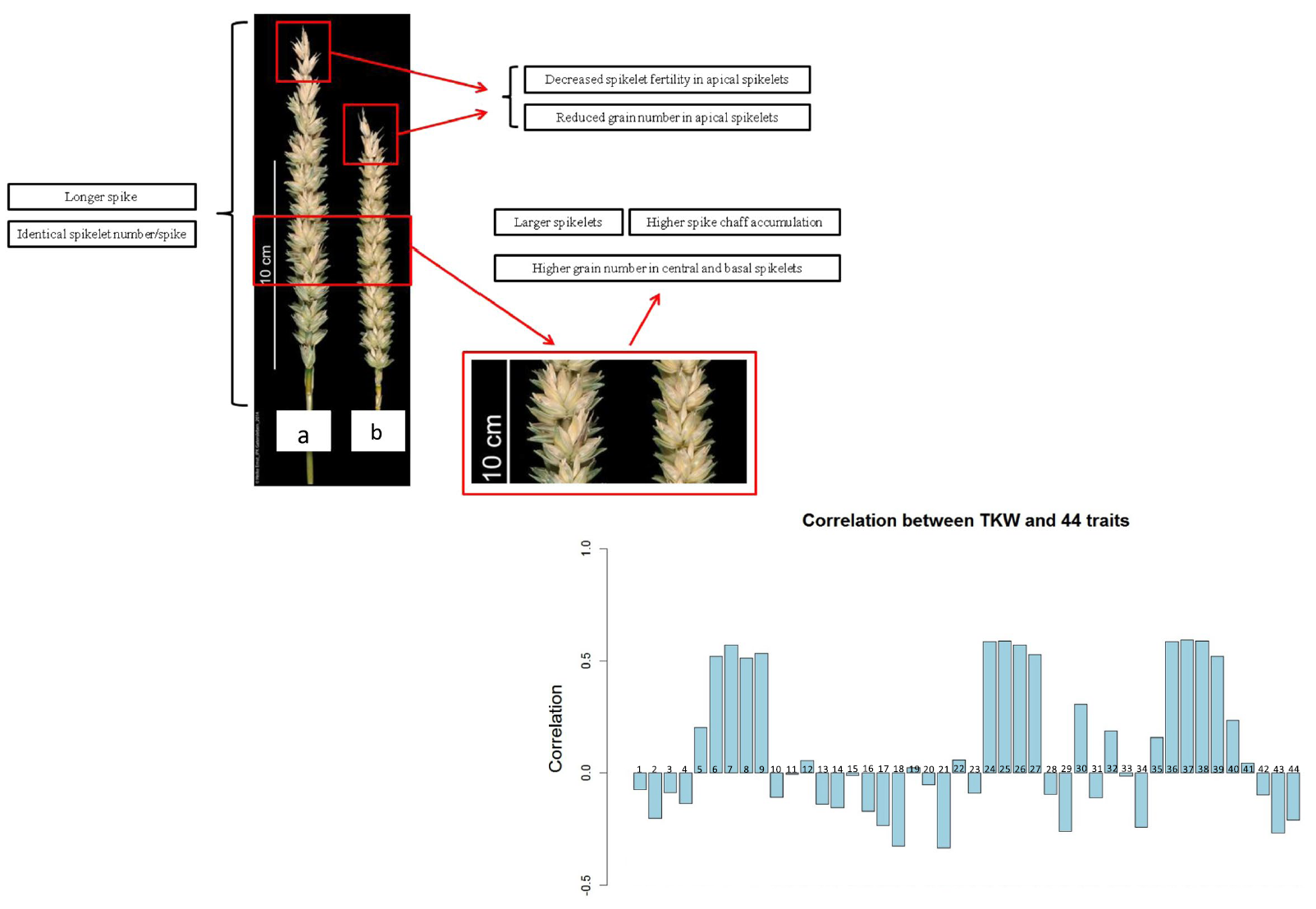
39. Manipulation and prediction of spike morphology traits for the improvement of grain yield in wheat.
Guo Z, Zhao Y, Röder MS, Reif JC, Ganal MW, Chen D, Schnurbusch T
Sci Rep. 2018 Sep 26;8(1):14435. doi: 10.1038/s41598-018-31977-3.
In wheat (Triticum spp.), modifying inflorescence (spike) morphology can increase grain number and size and thus improve yield. Here, we demonstrated the potential for manipulating and predicting spike morphology, based on 44 traits. In 12 wheat cultivars, we observed that detillering (removal of branches), which alters photosynthate distribution, changed spike morphology. Our genome-wide association study detected close associations between carbon partitioning (e.g. tiller number, main shoot dry weight) and spike morphology (e.g. spike length, spikelet density) traits in 210 cultivars. Most carbon-partitioning traits (e.g. tiller dry weight, harvest index) demonstrated high prediction abilities (>0.5). For spike morphology, some traits (e.g. total and fertile spikelet number, spike length) displayed high prediction abilities (0.3-0.5), but others (e.g. spikelet fertility, spikelet density) exhibited low prediction abilities (<0.2). Grain size traits were closely correlated in field and greenhouse experiments. Stepwise regression analysis suggests that significantly associated traits in the greenhouse explain 35.35% of the variation in grain yield and 67.63% of the variation in thousand-kernel weight in the field. Therefore, the traits identified in this study affect spike morphology; these traits can be used to predict and improve plant architecture and thus increase yield.
doi: 10.1038/s41598-018-31977-3 PubMed: 30258057 Google Scholar
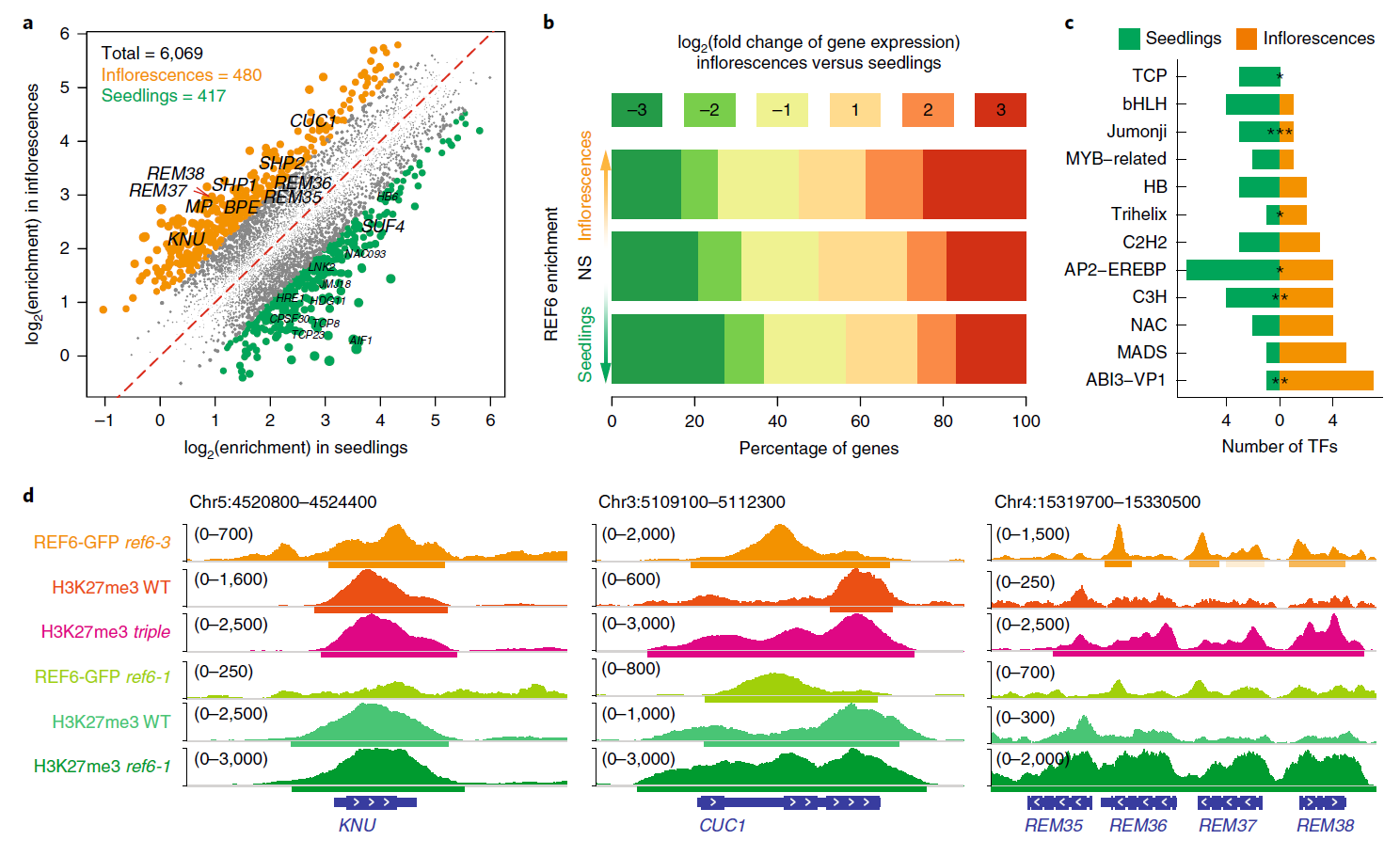
38. Dynamic and spatial restriction of Polycomb activity by plant histone demethylases.
Yan W, Chen D#, Smaczniak C, Engelhorn J, Liu H, Yang W, Graf A, Carles CC, Zhou DX, Kaufmann K
Nat Plants. 2018 Aug 13. doi: 10.1038/s41477-018-0219-5.
Targeted changes in chromatin state at thousands of genes are central to eukaryotic development. RELATIVE OF EARLY FLOWERING 6 (REF6) is a Jumonji-type histone demethylase that counteracts Polycomb repressive complex 2 (PRC2)-mediated gene silencing in plants and was reported to select its binding sites in a direct, sequence-specific manner1-3. Here we show that REF6 and its two close paralogues determine spatial 'boundaries' of the repressive histone H3K27me3 mark in the genome and control the tissue-specific release from PRC2-mediated gene repression. Targeted mutagenesis revealed that these histone demethylases display pleiotropic, redundant functions in plant development, several of which depend on trans factor-mediated recruitment. Thus, Jumonji-type histone demethylases restrict repressive chromatin domains and contribute to tissue-specific gene activation via complementary targeting mechanisms.
doi: 10.1038/s41477-018-0219-5 PubMed: 30104650 Google Scholar
37. A practical guide for DNase-seq data analysis: from data management to common applications.
Liu Y, Fu L, Kaufmann K, Chen D*, Chen M
Brief Bioinform. 2018 Jul 12. doi: 10.1093/bib/bby057.
Deoxyribonuclease I (DNase I)-hypersensitive site sequencing (DNase-seq) has been widely used to determine chromatin accessibility and its underlying regulatory lexicon. However, exploring DNase-seq data requires sophisticated downstream bioinformatics analyses. In this study, we first review computational methods for all of the major steps in DNase-seq data analysis, including experimental design, quality control, read alignment, peak calling, annotation of cis-regulatory elements, genomic footprinting and visualization. The challenges associated with each step are highlighted. Next, we provide a practical guideline and a computational pipeline for DNase-seq data analysis by integrating some of these tools. We also discuss the competing techniques and the potential applications of this pipeline for the analysis of analogous experimental data. Finally, we discuss the integration of DNase-seq with other functional genomics techniques.

36. Genetic dissection of pre-anthesis sub-phase durations during the reproductive spike development of wheat.
Guo Z, Chen D, Röder MS, Ganal MW, Schnurbusch T
Plant J. 2018 Jun 15. doi: 10.1111/tpj.13998.
Flowering time is an important factor affecting grain yield in wheat. In this study, we divided reproductive spike development into eight sub-phases. These sub-phases have the potential to be delicately manipulated to increase grain yield. We measured 36 traits with regard to sub-phase durations, determined three grain yield-related traits in eight field environments and mapped 15 696 single nucleotide polymorphism (SNP, based on 90k Infinium chip and 35k Affymetrix chip) markers in 210 wheat genotypes. Phenotypic and genetic associations between grain yield traits and sub-phase durations showed significant consistency (Mantel test; r = 0.5377, P < 0.001). The shared quantitative trait loci (QTLs) revealed by the genome-wide association study suggested a close association between grain yield and sub-phase duration, which may be attributed to effects on spikelet initiation/spikelet number (double ridge to terminal spikelet stage, DR-TS) and assimilate accumulation (green anther to anthesis stage, GA-AN). Moreover, we observed that the photoperiod-sensitivity allele at the Ppd-D1 locus on chromosome 2D markedly extended all sub-phase durations, which may contribute to its positive effects on grain yield traits. The dwarfing allele at the Rht-D1 (chromosome 4D) locus altered the sub-phase duration and displayed positive effects on grain yield traits. Data for 30 selected genotypes (from among the original 210 genotypes) in the field displayed a close association with that from the greenhouse. Most importantly, this study demonstrated specific connections to grain yield in narrower time windows (i.e. the eight sub-phases), rather than the entire stem elongation phase as a whole.
35. Plant and Floret Growth at Distinct Developmental Stages During the Stem Elongation Phase in Wheat.
Guo Z, Chen D, Schnurbusch T
Front Plant Sci. 2018 Mar 15;9:330. doi: 10.3389/fpls.2018.00330.
Floret development is critical for grain setting in wheat (Triticum aestivum), but more than 50% of grain yield potential (based on the maximum number of floret primordia) is lost during the stem elongation phase (SEP, from the terminal spikelet stage to anthesis). Dynamic plant (e.g., leaf area, plant height) and floret (e.g., anther and ovary size) growth and its connection with grain yield traits (e.g., grain number and width) are not clearly understood. In this study, for the first time, we dissected the SEP into seven stages to investigate plant (first experiment) and floret (second experiment) growth in greenhouse- and field-grown wheat. In the first experiment, the values of various plant growth trait indices at different stages were generally consistent between field and greenhouse and were independent of the environment. However, at specific stages, some traits significantly differed between the two environments. In the second experiment, phenotypic and genotypic similarity analysis revealed that grain number and size corresponded closely to ovary size at anthesis, suggesting that ovary size is strongly associated with grain number and size. Moreover, principal component analysis (PCA) showed that the top six principal components PCs explained 99.13, 98.61, 98.41, 98.35, and 97.93% of the total phenotypic variation at the green anther, yellow anther, tipping, heading, and anthesis stages, respectively. The cumulative variance explained by the first PC decreased with floret growth, with the highest value detected at the green anther stage (88.8%) and the lowest at the anthesis (50.09%). Finally, ovary size at anthesis was greater in wheat accessions with early release years than in accessions with late release years, and anther/ovary size shared closer connections with grain number/size traits at the late vs. early stages of floral development. Our findings shed light on the dynamic changes in plant and floret growth-related traits in wheat and the effects of the environment on these traits.
doi: 10.3389/fpls.2018.00330 PubMed: 29599792 Google Scholar
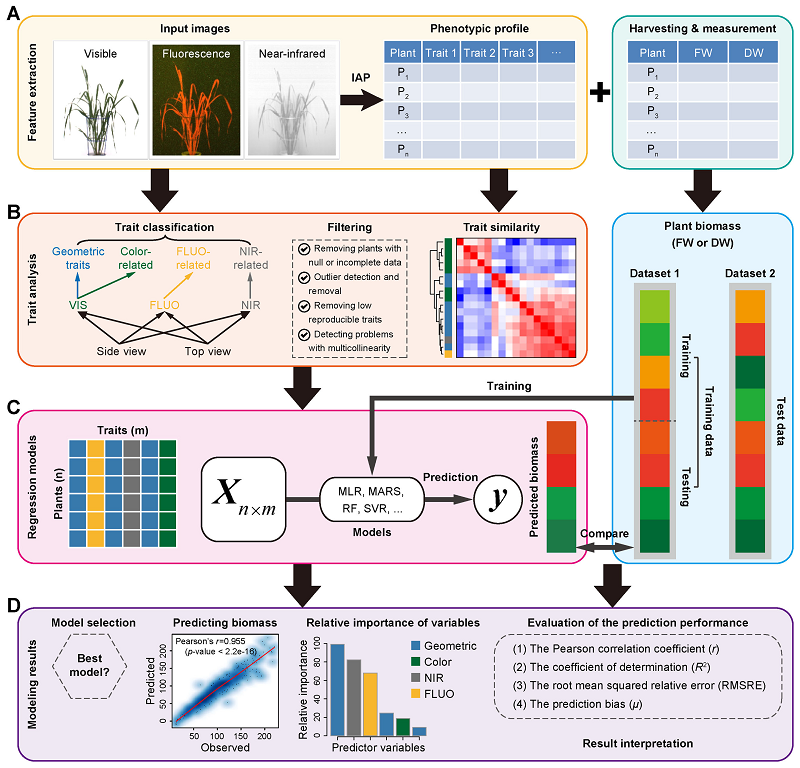
34. Predicting plant biomass accumulation from image-derived parameters.
Chen D*, Shi R, Pape JM, Neumann K, Arend D, Graner A, Chen M, Klukas C
Gigascience. 2018 Feb 1;7(2). doi: 10.1093/gigascience/giy001.
Background: Image-based high-throughput phenotyping technologies have been rapidly developed in plant science recently, and they provide a great potential to gain more valuable information than traditionally destructive methods. Predicting plant biomass is regarded as a key purpose for plant breeders and ecologists. However, it is a great challenge to find a predictive biomass model across experiments. Results: In the present study, we constructed 4 predictive models to examine the quantitative relationship between image-based features and plant biomass accumulation. Our methodology has been applied to 3 consecutive barley (Hordeum vulgare) experiments with control and stress treatments. The results proved that plant biomass can be accurately predicted from image-based parameters using a random forest model. The high prediction accuracy based on this model will contribute to relieving the phenotyping bottleneck in biomass measurement in breeding applications. The prediction performance is still relatively high across experiments under similar conditions. The relative contribution of individual features for predicting biomass was further quantified, revealing new insights into the phenotypic determinants of the plant biomass outcome. Furthermore, methods could also be used to determine the most important image-based features related to plant biomass accumulation, which would be promising for subsequent genetic mapping to uncover the genetic basis of biomass. Conclusions: We have developed quantitative models to accurately predict plant biomass accumulation from image data. We anticipate that the analysis results will be useful to advance our views of the phenotypic determinants of plant biomass outcome, and the statistical methods can be broadly used for other plant species.
doi: 10.1093/gigascience/giy001 PubMed: 29346559 Google Scholar
2017
33. Differences in DNA Binding Specificity of Floral Homeotic Protein Complexes Predict Organ-Specific Target Genes.
Smaczniak C, Muiño JM, Chen D, Angenent GC, Kaufmann K
Plant Cell. 2017 Aug;29(8):1822-1835. doi: 10.1105/tpc.17.00145.
Floral organ identities in plants are specified by the combinatorial action of homeotic master regulatory transcription factors. However, how these factors achieve their regulatory specificities is still largely unclear. Genome-wide in vivo DNA binding data show that homeotic MADS domain proteins recognize partly distinct genomic regions, suggesting that DNA binding specificity contributes to functional differences of homeotic protein complexes. We used in vitro systematic evolution of ligands by exponential enrichment followed by high-throughput DNA sequencing (SELEX-seq) on several floral MADS domain protein homo- and heterodimers to measure their DNA binding specificities. We show that specification of reproductive organs is associated with distinct binding preferences of a complex formed by SEPALLATA3 and AGAMOUS. Binding specificity is further modulated by different binding site spacing preferences. Combination of SELEX-seq and genome-wide DNA binding data allows differentiation between targets in specification of reproductive versus perianth organs in the flower. We validate the importance of DNA binding specificity for organ-specific gene regulation by modulating promoter activity through targeted mutagenesis. Our study shows that intrafamily protein interactions affect DNA binding specificity of floral MADS domain proteins. Differential DNA binding of MADS domain protein complexes plays a role in the specificity of target gene regulation.
2016
32. Genome-wide association analyses of 54 traits identified multiple loci for the determination of floret fertility in wheat.
Guo Z, Chen D*, Alqudah AM, Röder MS, Ganal MW, Schnurbusch T
New Phytol. 2017 Apr;214(1):257-270. doi: 10.1111/nph.14342.
Increasing grain yield is still the main target of wheat breeding; yet today's wheat plants utilize less than half of their yield potential. Owing to the difficulty of determining grain yield potential in a large population, few genetic factors regulating floret fertility (i.e. the difference between grain yield potential and grain number) have been reported to date. In this study, we conducted a genome-wide association study (GWAS) by quantifying 54 traits (16 floret fertility traits and 38 traits for assimilate partitioning and spike morphology) in 210 European winter wheat accessions. The results of this GWAS experiment suggested potential associations between floret fertility, assimilate partitioning and spike morphology revealed by shared quantitative trait loci (QTLs). Several candidate genes involved in carbohydrate metabolism, phytohormones or floral development colocalized with such QTLs, thereby providing potential targets for selection. Based on our GWAS results we propose a genetic network underlying floret fertility and related traits, nominating determinants for improved yield performance.

31. Measures for interoperability of phenotypic data: minimum information requirements and formatting.
Ćwiek-Kupczyńska H, Altmann T, Arend D, Arnaud E, Chen D, Cornut G, Fiorani F, Frohmberg W, Junker A, Klukas C, Lange M, Mazurek C, Nafissi A, Neveu P, van Oeveren J, Pommier C, Poorter H, Rocca-Serra P, Sansone SA, Scholz U, van Schriek M, Seren Ü, Usadel B, Weise S, Kersey P, Krajewski P
Plant Methods. 2016 Nov 9;12:44.
BACKGROUND: Plant phenotypic data shrouds a wealth of information which, when accurately analysed and linked to other data types, brings to light the knowledge about the mechanisms of life. As phenotyping is a field of research comprising manifold, diverse and time-consuming experiments, the findings can be fostered by reusing and combining existing datasets. Their correct interpretation, and thus replicability, comparability and interoperability, is possible provided that the collected observations are equipped with an adequate set of metadata. So far there have been no common standards governing phenotypic data description, which hampered data exchange and reuse. RESULTS: In this paper we propose the guidelines for proper handling of the information about plant phenotyping experiments, in terms of both the recommended content of the description and its formatting. We provide a document called "Minimum Information About a Plant Phenotyping Experiment", which specifies what information about each experiment should be given, and a Phenotyping Configuration for the ISA-Tab format, which allows to practically organise this information within a dataset. We provide examples of ISA-Tab-formatted phenotypic data, and a general description of a few systems where the recommendations have been implemented. CONCLUSIONS: Acceptance of the rules described in this paper by the plant phenotyping community will help to achieve findable, accessible, interoperable and reusable data.
doi: 10.1186/s13007-016-0144-4 PubMed: 27843484 Google Scholar
30. Efficient multiplex mutagenesis by RNA-guided Cas9 and its use in the characterization of regulatory elements in the AGAMOUS gene.
Yan W, Chen D, Kaufmann K
Plant Methods. 2016 Apr 25;12:23. doi: 10.1186/s13007-016-0125-7.
BACKGROUND: The efficiency of multiplex editing in plants by the RNA-guided Cas9 system is limited by efficient introduction of its components into the genome and by their activity. The possibility of introducing large fragment deletions by RNA-guided Cas9 tool provides the potential to study the function of any DNA region of interest in its 'endogenous' environment. RESULTS: Here, an RNA-guided Cas9 system was optimized to enable efficient multiplex editing in Arabidopsis thaliana. We demonstrate the flexibility of our system for knockout of multiple genes, and to generate heritable large-fragment deletions in the genome. As a proof of concept, the function of part of the second intron of the flower development gene AGAMOUS in Arabidopsis was studied by generating a Cas9-free mutant plant line in which part of this intron was removed from the genome. Further analysis revealed that deletion of this intron fragment results 40 % decrease of AGAMOUS gene expression without changing the splicing of the gene which indicates that this regulatory region functions as an activator of AGAMOUS gene expression. CONCLUSIONS: Our modified RNA-guided Cas9 system offers a versatile tool for the functional dissection of coding and non-coding DNA sequences in plants.
doi: 10.1186/s13007-016-0125-7 PubMed: 27118985 Google Scholar
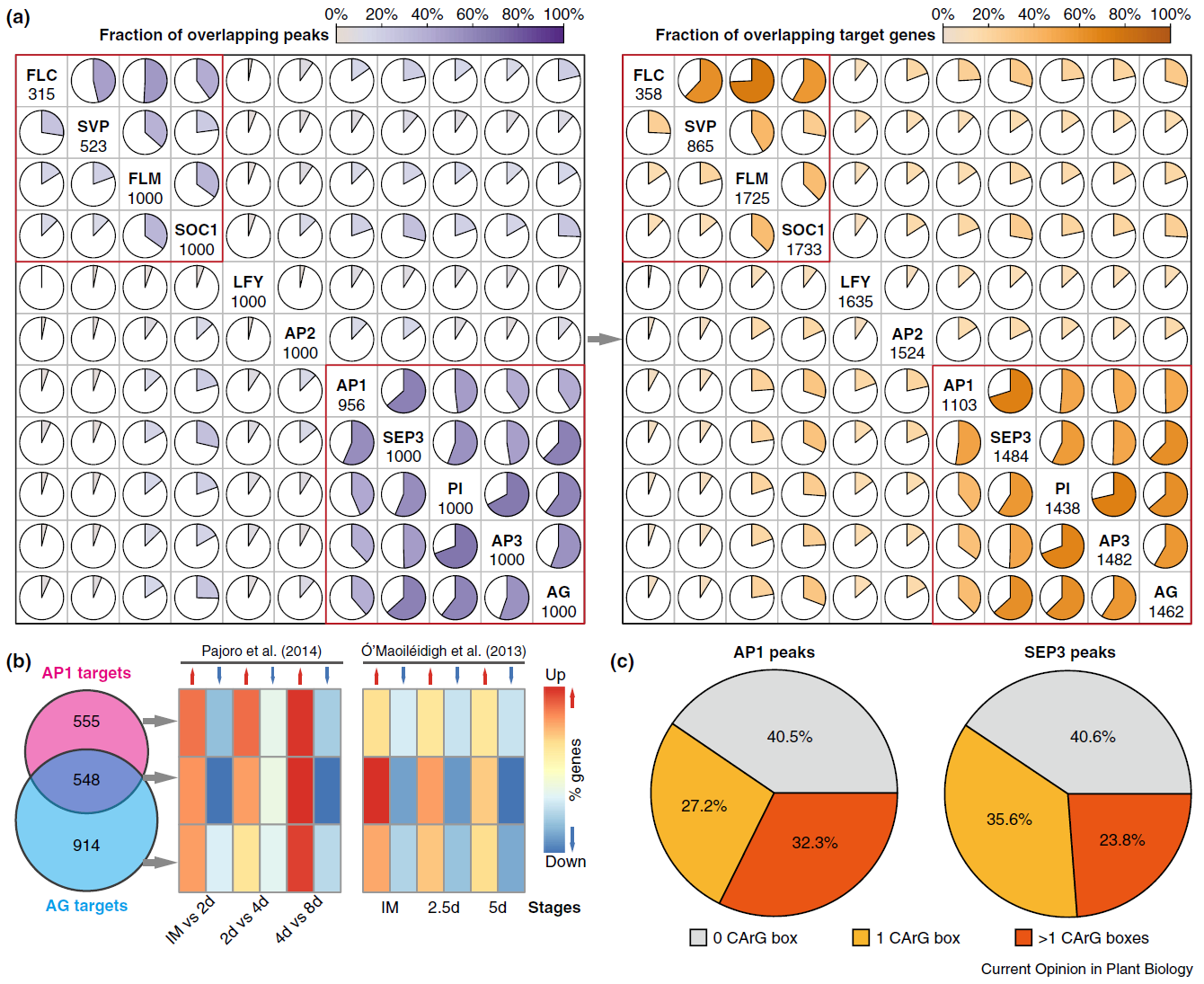
29. Molecular mechanisms of floral organ specification by MADS domain proteins.
Yan W, Chen D, Kaufmann K
Curr Opin Plant Biol. 2016 Feb;29:154-62. doi: 10.1016/j.pbi.2015.12.004.
Flower development is a model system to understand organ specification in plants. The identities of different types of floral organs are specified by homeotic MADS transcription factors that interact in a combinatorial fashion. Systematic identification of DNA-binding sites and target genes of these key regulators show that they have shared and unique sets of target genes. DNA binding by MADS proteins is not based on 'simple' recognition of a specific DNA sequence, but depends on DNA structure and combinatorial interactions. Homeotic MADS proteins regulate gene expression via alternative mechanisms, one of which may be to modulate chromatin structure and accessibility in their target gene promoters.
doi: 10.1016/j.pbi.2015.12.004 PubMed: 26802807 Google Scholar
2015
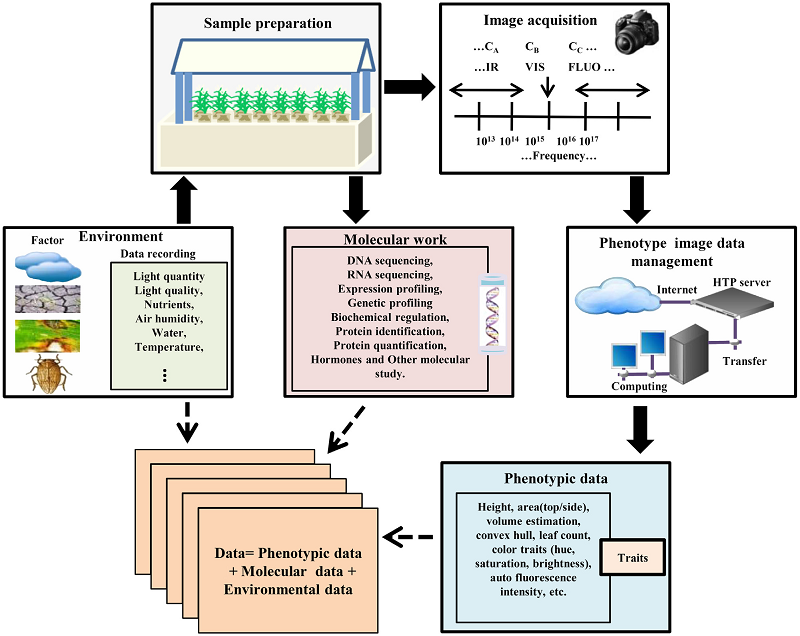
28. Advanced phenotyping and phenotype data analysis for the study of plant growth and development.
Rahaman MM, Chen D, Gillani Z, Klukas C, Chen M
Front Plant Sci. 2015 Aug 10;6:619. doi: 10.3389/fpls.2015.00619.
Due to an increase in the consumption of food, feed, fuel and to meet global food security needs for the rapidly growing human population, there is a necessity to breed high yielding crops that can adapt to the future climate changes, particularly in developing countries. To solve these global challenges, novel approaches are required to identify quantitative phenotypes and to explain the genetic basis of agriculturally important traits. These advances will facilitate the screening of germplasm with high performance characteristics in resource-limited environments. Recently, plant phenomics has offered and integrated a suite of new technologies, and we are on a path to improve the description of complex plant phenotypes. High-throughput phenotyping platforms have also been developed that capture phenotype data from plants in a non-destructive manner. In this review, we discuss recent developments of high-throughput plant phenotyping infrastructure including imaging techniques and corresponding principles for phenotype data analysis.
doi: 10.3389/fpls.2015.00619 PubMed: 26322060 Google Scholar

27. Towards recommendations for metadata and data handling in plant phenotyping.
Krajewski P, Chen D#, Ćwiek H, van Dijk AD, Fiorani F, Kersey P, Klukas C, Lange M, Markiewicz A, Nap JP, van Oeveren J, Pommier C, Scholz U, van Schriek M, Usadel B, Weise S
J Exp Bot. 2015 Sep;66(18):5417-27. doi: 10.1093/jxb/erv271.
Recent methodological developments in plant phenotyping, as well as the growing importance of its applications in plant science and breeding, are resulting in a fast accumulation of multidimensional data. There is great potential for expediting both discovery and application if these data are made publicly available for analysis. However, collection and storage of phenotypic observations is not yet sufficiently governed by standards that would ensure interoperability among data providers and precisely link specific phenotypes and associated genomic sequence information. This lack of standards is mainly a result of a large variability of phenotyping protocols, the multitude of phenotypic traits that are measured, and the dependence of these traits on the environment. This paper discusses the current situation of standardization in the area of phenomics, points out the problems and shortages, and presents the areas that would benefit from improvement in this field. In addition, the foundations of the work that could revise the situation are proposed, and practical solutions developed by the authors are introduced.
26. Genome-wide view of natural antisense transcripts in Arabidopsis thaliana.
Yuan C, Wang J, Harrison AP, Meng X, Chen D, Chen M
DNA Res. 2015 Jun;22(3):233-43. doi: 10.1093/dnares/dsv008.
Natural antisense transcripts (NATs) are endogenous transcripts that can form double-stranded RNA structures. Many protein-coding genes (PCs) and non-protein-coding genes (NPCs) tend to form cis-NATs and trans-NATs, respectively. In this work, we identified 4,080 cis-NATs and 2,491 trans-NATs genome-widely in Arabidopsis. Of these, 5,385 NAT-siRNAs were detected from the small RNA sequencing data. NAT-siRNAs are typically 21nt, and are processed by Dicer-like 1 (DCL1)/DCL2 and RDR6 and function in epigenetically activated situations, or 24nt, suggesting these are processed by DCL3 and RDR2 and function in environment stress. NAT-siRNAs are significantly derived from PC/PC pairs of trans-NATs and NPC/NPC pairs of cis-NATs. Furthermore, NAT pair genes typically have similar pattern of epigenetic status. Cis-NATs tend to be marked by euchromatic modifications, whereas trans-NATs tend to be marked by heterochromatic modifications.
25. Variance components, heritability and correlation analysis of anther and ovary size during the floral development of bread wheat.
Guo Z, Chen D, Schnurbusch T
J Exp Bot. 2015 Jun;66(11):3099-111. doi: 10.1093/jxb/erv117.
Anther and ovary development play an important role in grain setting, a crucial factor determining wheat (Triticum aestivum L.) yield. One aim of this study was to determine the heritability of anther and ovary size at different positions within a spikelet at seven floral developmental stages and conduct a variance components analysis. Relationships between anther and ovary size and other traits were also assessed. The thirty central European winter wheat genotypes used in this study were based on reduced height (Rht) and photoperiod sensitivity (Ppd) genes with variable genetic backgrounds. Identical experimental designs were conducted in a greenhouse and field simultaneously. Heritability of anther and ovary size indicated strong genetic control. Variance components analysis revealed that anther and ovary sizes of floret 3 (i.e. F3, the third floret from the spikelet base) and floret 4 (F4) were more sensitive to the environment compared with those in floret 1 (F1). Good correlations were found between spike dry weight and anther and ovary size in both greenhouse and field, suggesting that anther and ovary size are good predictors of each other, as well as spike dry weight in both conditions. Relationships between spike dry weight and anther and ovary size at F3/4 positions were stronger than at F1, suggesting that F3/4 anther and ovary size are better predictors of spike dry weight. Generally, ovary size showed a closer relationship with spike dry weight than anther size, suggesting that ovary size is a more reliable predictor of spike dry weight.
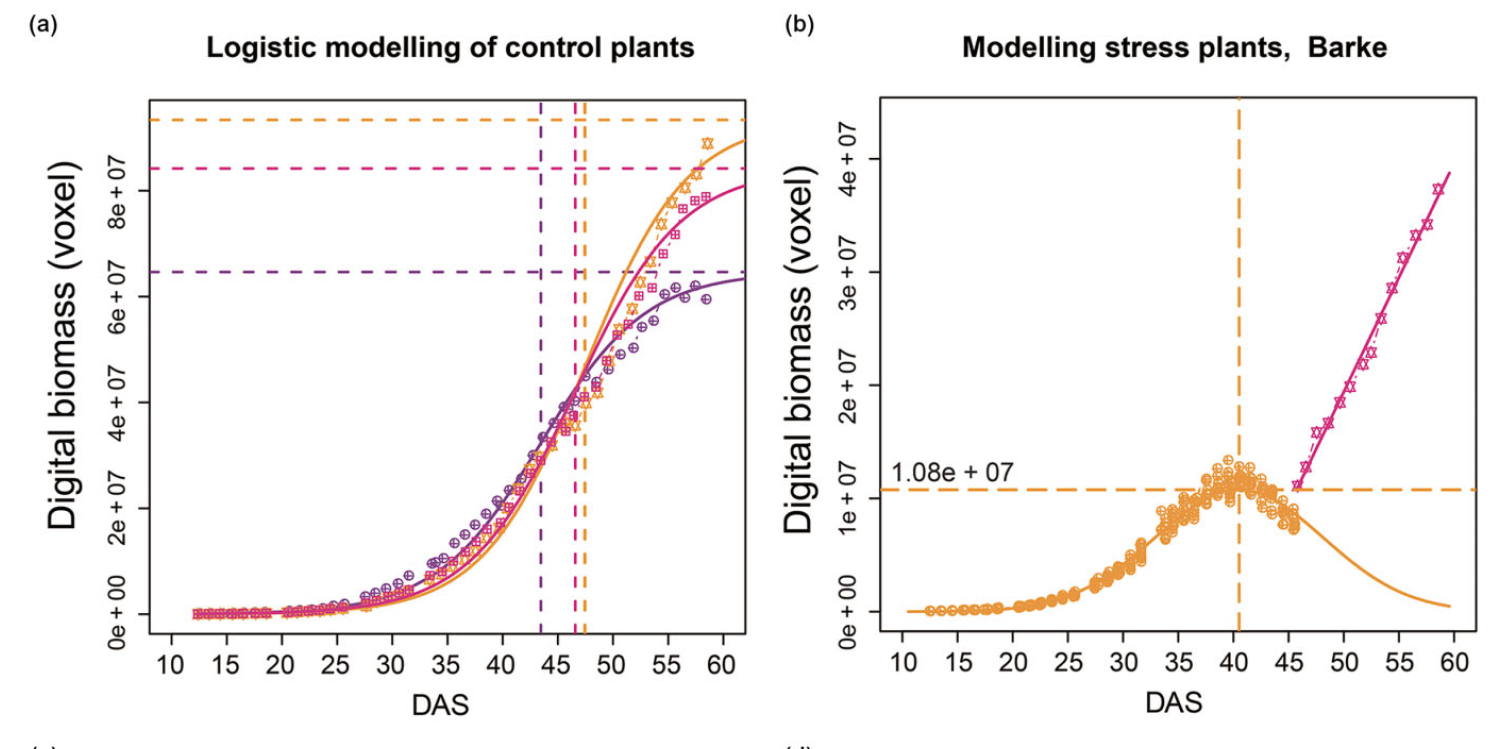
24. Dissecting spatiotemporal biomass accumulation in barley under different water regimes using high-throughput image analysis.
Neumann K, Klukas C, Friedel S, Rischbeck P, Chen D, Entzian A, Stein N, Graner A, Kilian B
Plant Cell Environ. 2015 Oct;38(10):1980-96. doi: 10.1111/pce.12516.
Phenotyping large numbers of genotypes still represents the rate-limiting step in many plant genetic experiments and in breeding. To address this issue, novel automated phenotyping technologies have been developed. We investigated for a core set of barley cultivars if high-throughput image analysis can help to dissect vegetative biomass accumulation in response to two different watering regimes under semi-controlled greenhouse conditions. We found that experiments, treatments, genotypes and genotype by environment interaction (G × E) can be characterized at any time point by certain digital traits. Biomass accumulation under control and stress conditions was highly heritable. Growth model-derived maximum vegetative biomass (K max), inflection point (I) and regrowth rate (k) were identified as promising candidate traits for genome-wide association studies. Drought stress symptoms can be visualized, dissected and modelled. Especially the highly heritable regrowth rate, which had the biggest influence on biomass accumulation in stress treatment, seems promising for future studies to improve drought tolerance in different crop species. A proof of concept study revealed potential correlations between digital traits obtained from pot experiments under greenhouse conditions and agronomic traits from field experiments. Overall, non-invasive, imaging-based phenotyping platforms under greenhouse conditions offer excellent possibilities for trait discovery, trait development and industrial applications.
2014

23. Dissecting the phenotypic components of crop plant growth and drought responses based on high-throughput image analysis.
Chen D, Neumann K, Friedel S, Kilian B, Chen M, Altmann T, Klukas C
Plant Cell. 2014 Dec;26(12):4636-55. doi: 10.1105/tpc.114.129601.
Significantly improved crop varieties are urgently needed to feed the rapidly growing human population under changing climates. While genome sequence information and excellent genomic tools are in place for major crop species, the systematic quantification of phenotypic traits or components thereof in a high-throughput fashion remains an enormous challenge. In order to help bridge the genotype to phenotype gap, we developed a comprehensive framework for high-throughput phenotype data analysis in plants, which enables the extraction of an extensive list of phenotypic traits from nondestructive plant imaging over time. As a proof of concept, we investigated the phenotypic components of the drought responses of 18 different barley (Hordeum vulgare) cultivars during vegetative growth. We analyzed dynamic properties of trait expression over growth time based on 54 representative phenotypic features. The data are highly valuable to understand plant development and to further quantify growth and crop performance features. We tested various growth models to predict plant biomass accumulation and identified several relevant parameters that support biological interpretation of plant growth and stress tolerance. These image-based traits and model-derived parameters are promising for subsequent genetic mapping to uncover the genetic basis of complex agronomic traits. Taken together, we anticipate that the analytical framework and analysis results presented here will be useful to advance our views of phenotypic trait components underlying plant development and their responses to environmental cues.
22. Genome-wide comparison of microRNAs and their targeted transcripts among leaf, flower and fruit of sweet orange.
Liu Y, Wang L, Chen D, Wu X, Huang D, Chen L, Li L, Deng X, Xu Q
BMC Genomics. 2014 Aug 20;15:695. doi: 10.1186/1471-2164-15-695.
BACKGROUND: In plants, microRNAs (miRNAs) regulate gene expression mainly at the post-transcriptional level. Previous studies have demonstrated that miRNA-mediated gene silencing pathways play vital roles in plant development. Here, we used a high-throughput sequencing approach to characterize the miRNAs and their targeted transcripts in the leaf, flower and fruit of sweet orange. RESULTS: A total of 183 known miRNAs and 38 novel miRNAs were identified. An in-house script was used to identify all potential secondary siRNAs derived from miRNA-targeted transcripts using sRNA and degradome sequencing data. Genome mapping revealed that these miRNAs were evenly distributed across the genome with several small clusters, and 69 pre-miRNAs were co-localized with simple sequence repeats (SSRs). Noticeably, the loop size of pre-miR396c was influenced by the repeat number of CUU unit. The expression pattern of miRNAs among different tissues and developmental stages were further investigated by both qRT-PCR and RNA gel blotting. Interestingly, Csi-miR164 was highly expressed in fruit ripening stage, and was validated to target a NAC transcription factor. This study depicts a global picture of miRNAs and their target genes in the genome of sweet orange, and focused on the comparison among leaf, flower and fruit tissues. CONCLUSIONS: This study provides a global view of miRNAs and their target genes in different tissue of sweet orange, and focused on the identification of miRNA involved in the regulation of fruit ripening. The results of this study lay a foundation for unraveling key regulators of orange fruit development and ripening on post-transcriptional level.
doi: 10.1186/1471-2164-15-695 PubMed: 25142253 Google Scholar
21. Prediction and functional analysis of the sweet orange protein-protein interaction network.
Ding YD, Chang JW, Guo J, Chen D, Li S, Xu Q, Deng XX, Cheng YJ, Chen LL
BMC Plant Biol. 2014 Aug 5;14:213. doi: 10.1186/s12870-014-0213-7.
BACKGROUND: Sweet orange (Citrus sinensis) is one of the most important fruits world-wide. Because it is a woody plant with a long growth cycle, genetic studies of sweet orange are lagging behind those of other species. RESULTS: In this analysis, we employed ortholog identification and domain combination methods to predict the protein-protein interaction (PPI) network for sweet orange. The K-nearest neighbors (KNN) classification method was used to verify and filter the network. The final predicted PPI network, CitrusNet, contained 8,195 proteins with 124,491 interactions. The quality of CitrusNet was evaluated using gene ontology (GO) and Mapman annotations, which confirmed the reliability of the network. In addition, we calculated the expression difference of interacting genes (EDI) in CitrusNet using RNA-seq data from four sweet orange tissues, and also analyzed the EDI distribution and variation in different sub-networks. CONCLUSIONS: Gene expression in CitrusNet has significant modular features. Target of rapamycin (TOR) protein served as the central node of the hormone-signaling sub-network. All evidence supported the idea that TOR can integrate various hormone signals and affect plant growth. CitrusNet provides valuable resources for the study of biological functions in sweet orange.
doi: 10.1186/s12870-014-0213-7 PubMed: 25091279 Google Scholar
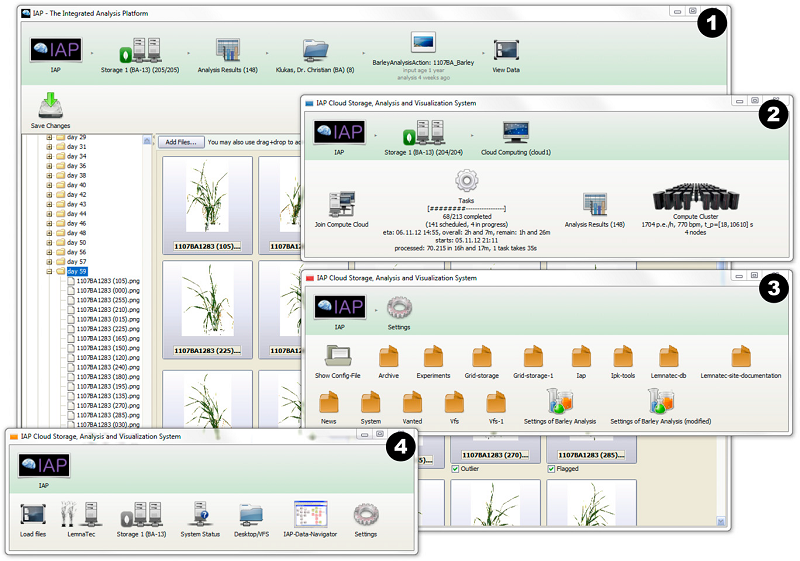
20. Integrated Analysis Platform: An Open-Source Information System for High-Throughput Plant Phenotyping.
Klukas C, Chen D, Pape JM
Plant Physiol. 2014 Jun;165(2):506-518.
High-throughput phenotyping is emerging as an important technology to dissect phenotypic components in plants. Efficient image processing and feature extraction are prerequisites to quantify plant growth and performance based on phenotypic traits. Issues include data management, image analysis, and result visualization of large-scale phenotypic data sets. Here, we present Integrated Analysis Platform (IAP), an open-source framework for high-throughput plant phenotyping. IAP provides user-friendly interfaces, and its core functions are highly adaptable. Our system supports image data transfer from different acquisition environments and large-scale image analysis for different plant species based on real-time imaging data obtained from different spectra. Due to the huge amount of data to manage, we utilized a common data structure for efficient storage and organization of data for both input data and result data. We implemented a block-based method for automated image processing to extract a representative list of plant phenotypic traits. We also provide tools for build-in data plotting and result export. For validation of IAP, we performed an example experiment that contains 33 maize (Zea mays 'Fernandez') plants, which were grown for 9 weeks in an automated greenhouse with nondestructive imaging. Subsequently, the image data were subjected to automated analysis with the maize pipeline implemented in our system. We found that the computed digital volume and number of leaves correlate with our manually measured data in high accuracy up to 0.98 and 0.95, respectively. In summary, IAP provides a multiple set of functionalities for import/export, management, and automated analysis of high-throughput plant phenotyping data, and its analysis results are highly reliable.
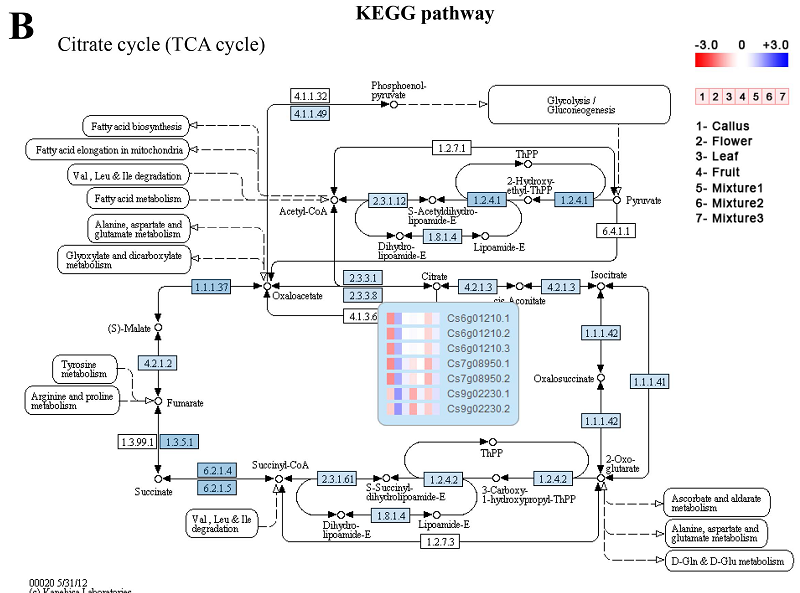
19. Citrus sinensis annotation project (CAP): a comprehensive database for sweet orange genome.
Wang J, Chen D#, Lei Y, Chang JW, Hao BH, Xing F, Li S, Xu Q, Deng XX, Chen LL
PLoS One. 2014 Jan 28;9(1):e87723. doi: 10.1371/journal.pone.0087723.
Citrus is one of the most important and widely grown fruit crop with global production ranking firstly among all the fruit crops in the world. Sweet orange accounts for more than half of the Citrus production both in fresh fruit and processed juice. We have sequenced the draft genome of a double-haploid sweet orange (C. sinensis cv. Valencia), and constructed the Citrus sinensis annotation project (CAP) to store and visualize the sequenced genomic and transcriptome data. CAP provides GBrowse-based organization of sweet orange genomic data, which integrates ab initio gene prediction, EST, RNA-seq and RNA-paired end tag (RNA-PET) evidence-based gene annotation. Furthermore, we provide a user-friendly web interface to show the predicted protein-protein interactions (PPIs) and metabolic pathways in sweet orange. CAP provides comprehensive information beneficial to the researchers of sweet orange and other woody plants, which is freely available at http://citrus.hzau.edu.cn/.
doi: 10.1371/journal.pone.0087723 PubMed: 24489955 Google Scholar
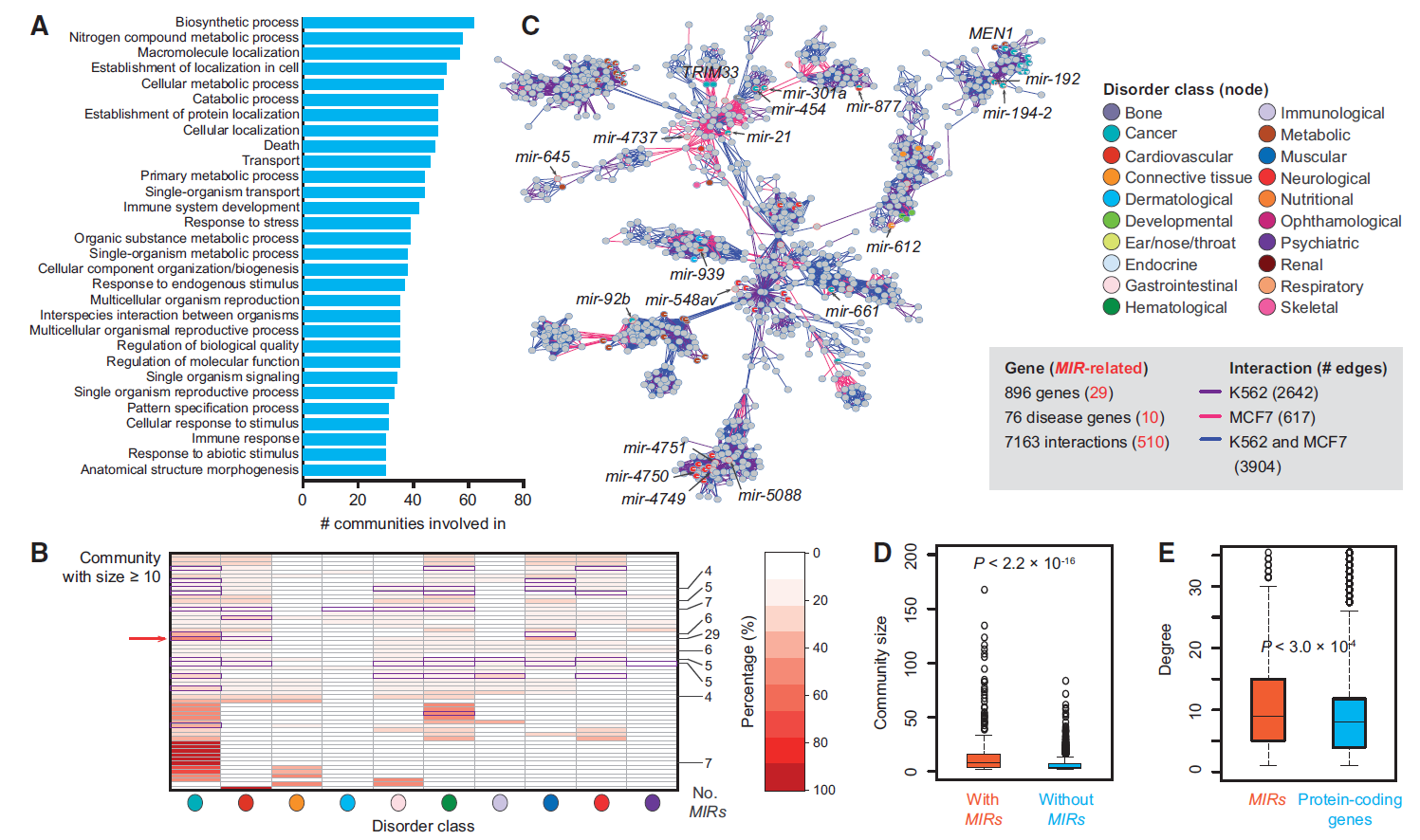
18. Dissecting the chromatin interactome of microRNA genes.
Chen D, Fu LY, Zhang Z, Li G, Zhang H, Jiang L, Harrison AP, Shanahan HP, Klukas C, Zhang HY, Ruan Y, Chen LL, Chen M
Nucleic Acids Res. 2014 Mar;42(5):3028-43. doi: 10.1093/nar/gkt1294.
Our knowledge of the role of higher-order chromatin structures in transcription of microRNA genes (MIRs) is evolving rapidly. Here we investigate the effect of 3D architecture of chromatin on the transcriptional regulation of MIRs. We demonstrate that MIRs have transcriptional features that are similar to protein-coding genes. RNA polymerase II-associated ChIA-PET data reveal that many groups of MIRs and protein-coding genes are organized into functionally compartmentalized chromatin communities and undergo coordinated expression when their genomic loci are spatially colocated. We observe that MIRs display widespread communication in those transcriptionally active communities. Moreover, miRNA-target interactions are significantly enriched among communities with functional homogeneity while depleted from the same community from which they originated, suggesting MIRs coordinating function-related pathways at posttranscriptional level. Further investigation demonstrates the existence of spatial MIR-MIR chromatin interacting networks. We show that groups of spatially coordinated MIRs are frequently from the same family and involved in the same disease category. The spatial interaction network possesses both common and cell-specific subnetwork modules that result from the spatial organization of chromatin within different cell types. Together, our study unveils an entirely unexplored layer of MIR regulation throughout the human genome that links the spatial coordination of MIRs to their co-expression and function.
2013
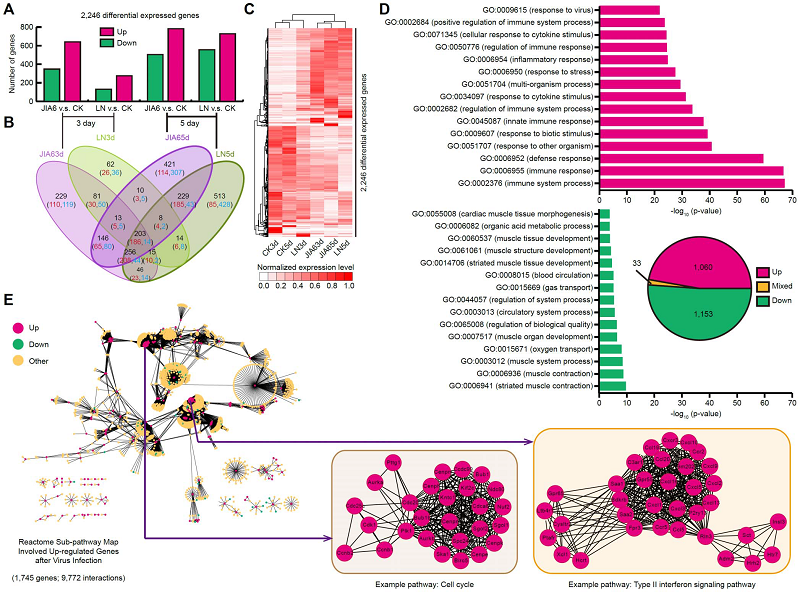
17. Insights into the increasing virulence of the swine-origin pandemic H1N1/2009 influenza virus.
Zou W, Chen D#, Xiong M, Zhu J, Lin X, Wang L, Zhang J, Chen L, Zhang H, Chen H, Chen M, Jin M
Sci Rep. 2013;3:1601. doi: 10.1038/srep01601.
Pandemic H1N1/2009 viruses have been stabilized in swine herds, and some strains display higher pathogenicity than the human-origin isolates. In this study, high-throughput RNA sequencing (RNA-seq) is applied to explore the systemic transcriptome responses of the mouse lungs infected by swine (Jia6/10) and human (LN/09) H1N1/2009 viruses. The transcriptome data show that Jia6/10 activates stronger virus-sensing signals, such as the toll-like receptor, RIG-I like receptor and NOD-like receptor signalings, as well as a stronger NF-κB and JAK-STAT signals, which play significant roles in inducing innate immunity. Most cytokines and interferon-stimulated genes show higher expression lever in Jia/06 infected groups. Meanwhile, virus Jia6/10 activates stronger production of reactive oxygen species, which might further promote higher mutation rate of the virus genome. Collectively, our data reveal that the swine-origin pandemic H1N1/2009 virus elicits a stronger innate immune reaction and pro-oxidation stimulation, which might relate closely to the increasing pathogenicity.
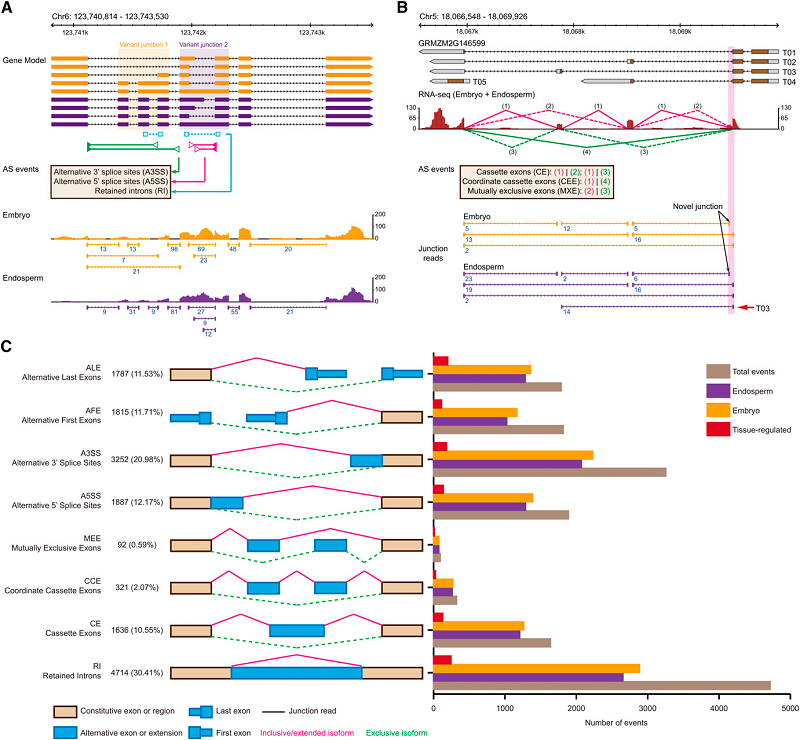
16. The differential transcription network between embryo and endosperm in the early developing maize seed.
Lu X, Chen D#, Shu D, Zhang Z, Wang W, Klukas C, Chen LL, Fan Y, Chen M, Zhang C
Plant Physiol. 2013 May;162(1):440-55. doi: 10.1104/pp.113.214874.
Transcriptome analysis of early-developing maize (Zea mays) seed was conducted using Illumina sequencing. We mapped 11,074,508 and 11,495,788 paired-end reads from endosperm and embryo, respectively, at 9 d after pollination to define gene structure and alternative splicing events as well as transcriptional regulators of gene expression to quantify transcript abundance in both embryo and endosperm. We identified a large number of novel transcribed regions that did not fall within maize annotated regions, and many of the novel transcribed regions were tissue-specifically expressed. We found that 50.7% (8,556 of 16,878) of multiexonic genes were alternatively spliced, and some transcript isoforms were specifically expressed either in endosperm or in embryo. In addition, a total of 46 trans-splicing events, with nine intrachromosomal events and 37 interchromosomal events, were found in our data set. Many metabolic activities were specifically assigned to endosperm and embryo, such as starch biosynthesis in endosperm and lipid biosynthesis in embryo. Finally, a number of transcription factors and imprinting genes were found to be specifically expressed in embryo or endosperm. This data set will aid in understanding how embryo/endosperm development in maize is differentially regulated.
2012
15. The draft genome of sweet orange (Citrus sinensis).
Xu Q, Chen LL, Ruan X, Chen D, Zhu A, Chen C, Bertrand D, Jiao WB, Hao BH, Lyon MP, Chen J, Gao S, Xing F, Lan H, Chang JW, Ge X, Lei Y, Hu Q, Miao Y, Wang L, Xiao S, Biswas MK, Zeng W, Guo F, Cao H, Yang X, Xu XW, Cheng YJ, Xu J, Liu JH, Luo OJ, Tang Z, Guo WW, Kuang H, Zhang HY, Roose ML, Nagarajan N, Deng XX, Ruan Y
Nat Genet. 2013 Jan;45(1):59-66. doi: 10.1038/ng.2472.
Oranges are an important nutritional source for human health and have immense economic value. Here we present a comprehensive analysis of the draft genome of sweet orange (Citrus sinensis). The assembled sequence covers 87.3% of the estimated orange genome, which is relatively compact, as 20% is composed of repetitive elements. We predicted 29,445 protein-coding genes, half of which are in the heterozygous state. With additional sequencing of two more citrus species and comparative analyses of seven citrus genomes, we present evidence to suggest that sweet orange originated from a backcross hybrid between pummelo and mandarin. Focused analysis on genes involved in vitamin C metabolism showed that GalUR, encoding the rate-limiting enzyme of the galacturonate pathway, is significantly upregulated in orange fruit, and the recent expansion of this gene family may provide a genomic basis. This draft genome represents a valuable resource for understanding and improving many important citrus traits in the future.
14. Pseudorabies virus infected porcine epithelial cell line generates a diverse set of host microRNAs and a special cluster of viral microRNAs.
Wu YQ, Chen D#, He HB, Chen DS, Chen LL, Chen HC, Liu ZF
PLoS One. 2012;7(1):e30988. doi: 10.1371/journal.pone.0030988.
Pseudorabies virus (PRV) belongs to Alphaherpesvirinae subfamily that causes huge economic loss in pig industry worldwide. It has been recently demonstrated that many herpesviruses encode microRNAs (miRNAs), which play crucial roles in viral life cycle. However, the knowledge about PRV-encoded miRNAs is still limited. Here, we report a comprehensive analysis of both viral and host miRNA expression profiles in PRV-infected porcine epithelial cell line (PK-15). Deep sequencing data showed that the ∼4.6 kb intron of the large latency transcript (LLT) functions as a primary microRNA precursor (pri-miRNA) that encodes a cluster of 11 distinct miRNAs in the PRV genome, and 209 known and 39 novel porcine miRNAs were detected. Viral miRNAs were further confirmed by stem-loop RT-PCR and northern blot analysis. Intriguingly, all of these viral miRNAs exhibited terminal heterogeneity both at the 5' and 3' ends. Seven miRNA genes produced mature miRNAs from both arms and two of the viral miRNA genes showed partially overlapped in their precursor regions. Unexpectedly, a terminal loop-derived small RNA with high abundance and one special miRNA offset RNA (moRNA) were processed from a same viral miRNA precursor. The polymorphisms of viral miRNAs shed light on the complexity of host miRNA-processing machinery and viral miRNA-regulatory mechanism. The swine genes and PRV genes were collected for target prediction of the viral miRNAs, revealing a complex network formed by both host and viral genes. GO enrichment analysis of host target genes suggests that PRV miRNAs are involved in complex cellular pathways including cell death, immune system process, metabolic pathway, indicating that these miRNAs play significant roles in virus-cells interaction of PRV and its hosts. Collectively, these data suggest that PRV infected epithelial cell line generates a diverse set of host miRNAs and a special cluster of viral miRNAs, which might facilitate PRV replication in cells.
doi: 10.1371/journal.pone.0030988 PubMed: 22292087 Google Scholar
13. PlantNATsDB: a comprehensive database of plant natural antisense transcripts.
Chen D, Yuan C, Zhang J, Zhang Z, Bai L, Meng Y, Chen LL, Chen M
Nucleic Acids Res. 2012 Jan;40(Database issue):D1187-93. doi: 10.1093/nar/gkr823.
Natural antisense transcripts (NATs), as one type of regulatory RNAs, occur prevalently in plant genomes and play significant roles in physiological and pathological processes. Although their important biological functions have been reported widely, a comprehensive database is lacking up to now. Consequently, we constructed a plant NAT database (PlantNATsDB) involving approximately 2 million NAT pairs in 69 plant species. GO annotation and high-throughput small RNA sequencing data currently available were integrated to investigate the biological function of NATs. PlantNATsDB provides various user-friendly web interfaces to facilitate the presentation of NATs and an integrated, graphical network browser to display the complex networks formed by different NATs. Moreover, a 'Gene Set Analysis' module based on GO annotation was designed to dig out the statistical significantly overrepresented GO categories from the specific NAT network. PlantNATsDB is currently the most comprehensive resource of NATs in the plant kingdom, which can serve as a reference database to investigate the regulatory function of NATs. The PlantNATsDB is freely available at http://bis.zju.edu.cn/pnatdb/.
2011
12. MyBioNet: interactively visualize, edit and merge biological networks on the web.
Huang D, Huang Y, Bai Y, Chen D, Hofestädt R, Klukas C, Chen M
Bioinformatics. 2011 Dec 1;27(23):3321-2. doi: 10.1093/bioinformatics/btr557.
SUMMARY: MyBioNet is a web-based application for biological network analysis, which provides user-friendly web interfaces to visualize, edit and merge biological networks. In addition, MyBioNet integrated KEGG metabolic network data from 1366 organisms and allows users to search and navigate interesting networks. AVAILABILITY AND IMPLEMENTATION: All KEGG metabolic network data are organized and stored in the MySQL database. MyBioNet is implemented in Flex/Actionscript and PHP languages and deployed on an Apache web server. MyBioNet is accessible through all the Flash-embedded browsers at http://bis.zju.edu.cn/mybionet/. CONTACT: mchen@zju.edu.cn.
doi: 10.1093/bioinformatics/btr557 PubMed: 21984760 Google Scholar
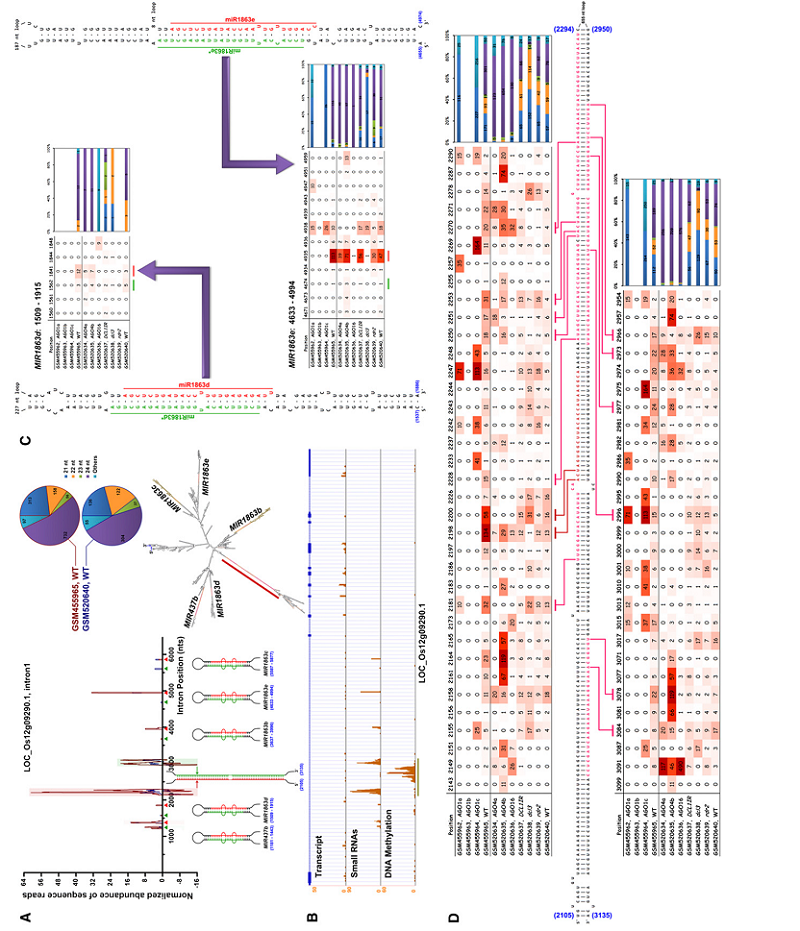
11. Plant siRNAs from introns mediate DNA methylation of host genes.
Chen D, Meng Y, Yuan C, Bai L, Huang D, Lv S, Wu P, Chen LL, Chen M
RNA. 2011 Jun;17(6):1012-24. doi: 10.1261/rna.2589011.
Small RNAs (sRNAs), largely known as microRNAs (miRNAs) and short interfering RNAs (siRNAs), emerged as the critical components of genetic and epigenetic regulation in eukaryotic genomes. In animals, a sizable portion of miRNAs reside within the introns of protein-coding genes, designated as mirtron genes. Recently, high-throughput sequencing (HTS) revealed a huge amount of sRNAs that derived from introns in plants, such as the monocot rice (Oryza sativa). However, the biogenesis and the biological functions of this kind of sRNAs remain elusive. Here, we performed a genome-scale survey of intron-derived sRNAs in rice based on HTS data. Several introns were found to have great potential to form internal hairpin structures, and the short hairpins could generate miRNAs while the larger ones could produce siRNAs. Furthermore, 22 introns, termed "sirtrons," were identified from the rice protein-coding genes. The single-stranded sirtrons produced a diverse set of siRNAs from long hairpin structures. These sirtron-derived siRNAs are dominantly 21 nt, 22 nt, and 24 nt in length, whose production relied on DCL4, DCL2, and DCL3, respectively. We also observed a strong tendency for the sirtron-derived siRNAs to be coexpressed with their host genes. Finally, the 24-nt siRNAs incorporated with Argonaute 4 (AGO4) could direct DNA methylation on their host genes. In this regard, homeostatic self-regulation between intron-derived siRNAs and their host genes was proposed.
2010
10. A web-based platform for rice microarray annotation and data analysis.
Chen D, Zhang F, Yuan C, Lu J, Li X, Chen M
Sci China Life Sci. 2010 Dec;53(12):1467-73. doi: 10.1007/s11427-010-4101-6.
Rice (Oryza sativa) feeds over half of the global population. A web-based integrated platform for rice microarray annotation and data analysis in various biological contexts is presented, which provides a convenient query for comprehensive annotation compared with similar databases. Coupled with existing rice microarray data, it provides online analysis methods from the perspective of bioinformatics. This comprehensive bioinformatics analysis platform is composed of five modules, including data retrieval, microarray annotation, sequence analysis, results visualization and data analysis. The BioChip module facilitates the retrieval of microarray data information via identifiers of "Probe Set ID", "Locus ID" and "Analysis Name". The BioAnno module is used to annotate the gene or probe set based on the gene function, the domain information, the KEGG biochemical and regulatory pathways and the potential microRNA which regulates the genes. The BioSeq module lists all of the related sequence information by a microarray probe set. The BioView module provides various visual results for the microarray data. The BioAnaly module is used to analyze the rice microarray's data set.
doi: 10.1007/s11427-010-4101-6 PubMed: 21181349 Google Scholar
09. RNA editing of nuclear transcripts in Arabidopsis thaliana.
Meng Y, Chen D, Jin Y, Mao C, Wu P, Chen M
BMC Genomics. 2010 Dec 2;11 Suppl 4:S12. doi: 10.1186/1471-2164-11-S4-S12.
BACKGROUND: RNA editing is a transcript-based layer of gene regulation. To date, no systemic study on RNA editing of plant nuclear genes has been reported. Here, a transcriptome-wide search for editing sites in nuclear transcripts of Arabidopsis (Arabidopsis thaliana) was performed. RESULTS: MPSS (massively parallel signature sequencing) and PARE (parallel analysis of RNA ends) data retrieved from public databases were utilized, focusing on one-base-conversion editing. Besides cytidine (C)-to-uridine (U) editing in mitochondrial transcripts, many nuclear transcripts were found to be diversely edited. Interestingly, a sizable portion of these nuclear genes are involved in chloroplast- or mitochondrion-related functions, and many editing events are tissue-specific. Some editing sites, such as adenosine (A)-to-U editing loci, were found to be surrounded by peculiar elements. The editing events of some nuclear transcripts are highly enriched surrounding the borders between coding sequences (CDSs) and 3' untranslated regions (UTRs), suggesting site-specific editing. Furthermore, RNA editing is potentially implicated in new start or stop codon generation, and may affect alternative splicing of certain protein-coding transcripts. RNA editing in the precursor microRNAs (pre-miRNAs) of ath-miR854 family, resulting in secondary structure transformation, implies its potential role in microRNA (miRNA) maturation. CONCLUSIONS: To our knowledge, the results provide the first global view of RNA editing in plant nuclear transcripts.
doi: 10.1186/1471-2164-11-S4-S12 PubMed: 21143795 Google Scholar
08. Functional characterization of plant small RNAs based on next-generation sequencing data.
Chen M, Meng Y, Gu H, Chen D
Comput Biol Chem. 2010 Dec;34(5-6):308-12. doi: 10.1016/j.compbiolchem.2010.10.001.
The currently developed next-generation sequencing (NGS) technology has significantly enhanced our capacity of small RNA (sRNA) exploration. Several ambitious sRNA projects have been established based on the technical support of NGS. Thanks to the high-throughput feature of NGS, huge amounts of sRNA sequencing data have been generated. However, much more research efforts are needed to further exploit these valuable data. In this study, we carried out functional analyses of sRNAs from 26 angiosperms by utilizing public sRNA NGS data. We proposed that the endogenous sRNAs largely represented by the 24-nt ones had a potential role in transposable element (TE) control in both Arabidopsis (Arabidopsis thaliana) and rice (Oryza sativa), based on the comparison of sRNA locus densities within the TEs to the non-TE genes. Functional analysis was performed for the predicted targets of the conserved sRNAs that were classified into eudicot-specific, monocot-specific, and angiosperm-conserved ones. Moreover, several miRNA families were found to be highly conserved, and miR396 was suggested to be the most conserved miRNA family between the eudicots and the monocots, indicating its essential role in angiosperm development. At last, we demonstrated that it was a great challenge for researchers to fully exploit these huge sRNA NGS datasets and numerous sRNA species remained to be uncovered and functionally characterized.
doi: 10.1016/j.compbiolchem.2010.10.001 PubMed: 21030312 Google Scholar
07. PmiRKB: a plant microRNA knowledge base.
Meng Y, Gou L, Chen D, Mao C, Jin Y, Wu P, Chen M
Nucleic Acids Res. 2011 Jan;39(Database issue):D181-7. doi: 10.1093/nar/gkq721.
MicroRNAs (miRNAs), one type of small RNAs (sRNAs) in plants, play an essential role in gene regulation. Several miRNA databases were established; however, successively generated new datasets need to be collected, organized and analyzed. To this end, we have constructed a plant miRNA knowledge base (PmiRKB) that provides four major functional modules. In the 'SNP' module, single nucleotide polymorphism (SNP) data of seven Arabidopsis (Arabidopsis thaliana) accessions and 21 rice (Oryza sativa) subspecies were collected to inspect the SNPs within pre-miRNAs (precursor microRNAs) and miRNA-target RNA duplexes. Depending on their locations, SNPs can affect the secondary structures of pre-miRNAs, or interactions between miRNAs and their targets. A second module, 'Pri-miR', can be used to investigate the tissue-specific, transcriptional contexts of pre- and pri-miRNAs (primary microRNAs), based on massively parallel signature sequencing data. The third module, 'MiR-Tar', was designed to validate thousands of miRNA-target pairs by using parallel analysis of RNA end (PARE) data. Correspondingly, the fourth module, 'Self-reg', also used PARE data to investigate the metabolism of miRNA precursors, including precursor processing and miRNA- or miRNA*-mediated self-regulation effects on their host precursors. PmiRKB can be freely accessed at http://bis.zju.edu.cn/pmirkb/.
06. High-throughput degradome sequencing can be used to gain insights into microRNA precursor metabolism.
Meng Y, Gou L, Chen D, Wu P, Chen M
J Exp Bot. 2010 Sep;61(14):3833-7. doi: 10.1093/jxb/erq209.
The approximately 21 nucleotide microRNAs (miRNAs) are one type of well-defined small RNA species, and they play critical roles in various biological processes in organisms. In plants, most miRNAs exert repressive regulation on their targets through cleavage, and a number of miRNA-target pairs have been validated either by modified 5' RACE (rapid amplification of cDNA ends), or by newly developed high-throughput strategies. All these data have greatly advanced our understanding of the regulatory roles of plant miRNAs. On the other hand, deep insights into miRNA precursor processing, and miRNA- or miRNA*-mediated self-regulation of their host precursors could be gained from high-throughput degradome sequencing data, based on the general framework of miRNA generation in plants. Here, the focus is on the recent research progress on this issue, and several interesting points were raised.
05. Methodological framework for functional characterization of plant microRNAs.
Chen M, Meng Y, Mao C, Chen D, Wu P
J Exp Bot. 2010 May;61(9):2271-80. doi: 10.1093/jxb/erq087.
Since the beginning of this century, microRNAs (miRNAs), which are tiny RNA molecules, have become one of the major research topics on gene expression regulation in both animals and plants. The major task of miRNA study is to elucidate how the miRNAs are expressed in vivo, how they exert regulatory effects on their targets, and how they can be qualitatively or quantitatively cloned. For these purposes, the methodology of miRNA study has been developed and significantly improved in recent years. The focus here is on a number of powerful methods for plant miRNA research including bioinformatics tools and experimental approaches being used for upstream or downstream analysis of miRNAs or miRNA cloning. Some discrepancies exist in the miRNA research methodology between plants and animals, for example, 5' modified RACE (Rapid Amplification of cDNA Ends) can be used for cleavage target validation only in plants. However, numerous common methods are shared by these two miRNA research areas. Thus, this review will enhance our understanding of miRNA research methodology in organisms.
04. Small RNAs in angiosperms: sequence characteristics, distribution and generation.
Chen D, Meng Y, Ma X, Mao C, Bai Y, Cao J, Gu H, Wu P, Chen M
Bioinformatics. 2010 Jun 1;26(11):1391-4. doi: 10.1093/bioinformatics/btq150.
High-throughput sequencing (HTS) has opened up a new era for small RNA (sRNA) exploration. Using HTS data for a global survey of sRNAs in 26 angiosperms, elevated GC contents were detected in the monocots, whereas the 5(')-terminal compositions were quite uniform among the angiosperms. Chromosome-wide distribution patterns of sRNAs were investigated by using scrolling-window analysis. We performed de novo natural antisense transcript (NAT) prediction, and found that the overlapping regions of trans-NATs, but not cis-NATs, were hotspots for sRNA generation. One cis-NAT generates phased natural antisense short interfering RNAs (nat-siRNAs) specifically from flowers in Arabidopsis, while one in rice produces phased nat-siRNAs from grains, suggesting their organ-specific regulatory roles.
doi: 10.1093/bioinformatics/btq150 PubMed: 20378553 Google Scholar
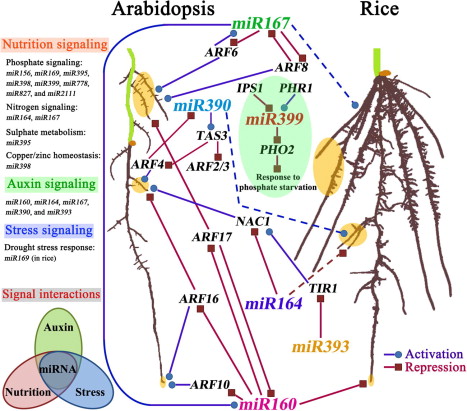
03. MicroRNA-mediated signaling involved in plant root development.
Meng Y, Ma X, Chen D, Wu P, Chen M
Biochem Biophys Res Commun. 2010 Mar 12;393(3):345-9. doi: 10.1016/j.bbrc.2010.01.129.
MicroRNA (miRNA), recently recognized as a critical post-transcriptional modulator of gene expression, is involved in numerous biological processes in both animals and plants. Although eudicots and monocots, such as the model plants Arabidopsis (Arabidopsis thaliana) and rice (Oryza sativa), possess distinct root systems, several homologous miRNA families are reported to be involved in root growth control in both plants. Consistent with recent notion that numerous signaling pathways are implicated in root development, these miRNAs are implicated in auxin signaling, nutrition metabolism, or stress response and have potential role in mediating the signal interactions. However, a recapitulative representation of these results is especially desired. This review provides a global view of the involvement of miRNAs in root development focusing on the two plants, Arabidopsis and rice. Based on current research advances, several innovative mechanisms of miRNA transcription, feedback regulatory circuit between miRNAs and transcription factors (TFs), and miRNA-mediated signal interactions are also discussed.
doi: 10.1016/j.bbrc.2010.01.129 PubMed: 20138828 Google Scholar
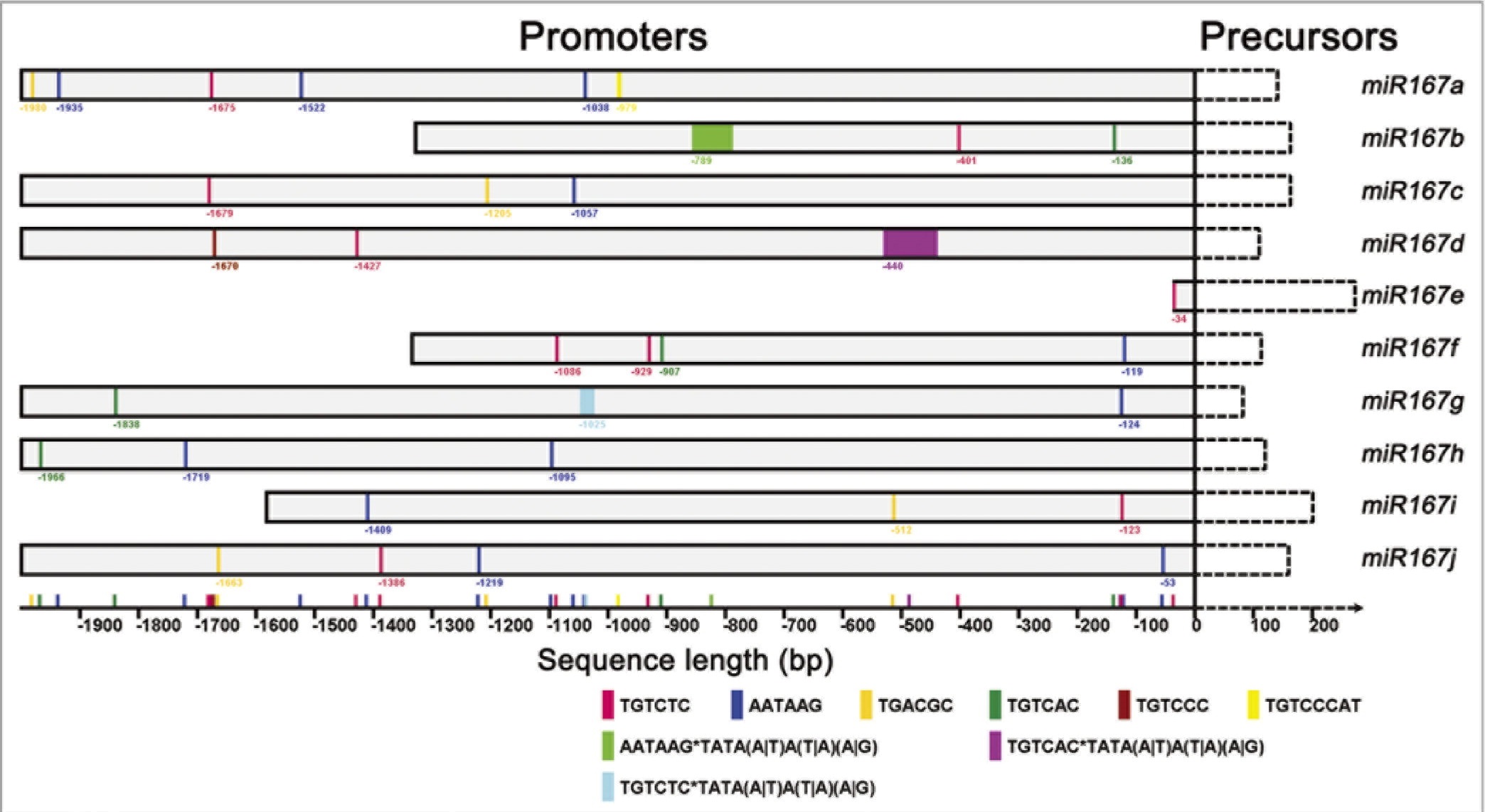
02. Mechanisms of microRNA-mediated auxin signaling inferred from the rice mutant osaxr.
Meng Y, Chen D, Ma X, Mao C, Cao J, Wu P, Chen M
Plant Signal Behav. 2010 Mar;5(3):252-4.
Auxin, known as the central hormone, plays essential roles in plant growth and development. In auxin signaling pathways, the tiny RNA molecules, i.e., microRNAs (miRNAs), show their strong potential in modulating the auxin signal transduction. Recently, we isolated a novel auxin resistant rice mutant osaxr (Oryza sativa auxin resistant) that exhibited plethoric root defects. Microarray experiments were carried out to investigate the expression patterns of both the miRNAs and the protein-coding genes in osaxr. A number of miRNAs showed reduced auxin sensitivity in osaxr compared with the wild type (WT), which may contribute to the auxin-resistant phenotype of the mutant. Auxin response elements (AuxREs) were demonstrated to be more frequently present in the promoters of auxin-related miRNAs. In our previous report, a comparative analysis of miRNA and protein-coding gene expression datasets uncovered a number of reciprocally expressed miRNA-target pairs. A feedback circuit between miRNA and auxin response factor (ARF) was then proposed. Here, we will discuss in-depth some points raised in the previous report, in particular, the organ-specific expression patterns of miR164, the feedback regulatory model between miR167 and certain ARFs, and the potential signal interactions between auxin and nutrition or stress that are mediated by miRNAs in rice roots.
2009

01. Genome-wide survey of rice microRNAs and microRNA-target pairs in the root of a novel auxin-resistant mutant.
Meng Y, Huang F, Shi Q, Cao J, Chen D, Zhang J, Ni J, Wu P, Chen M
Planta. 2009 Oct;230(5):883-98. doi: 10.1007/s00425-009-0994-3.
Auxin is one of the central hormones in plants, and auxin response factor (ARF) is a key regulator in the early auxin response. MicroRNAs (miRNAs) play an essential role in auxin signal transduction, but knowledge remains limited about the regulatory network between miRNAs and protein-coding genes (e.g. ARFs) involved in auxin signalling. In this study, we used a novel auxin-resistant rice mutant with plethoric root defects to investigate the miRNA expression patterns using microarray analysis. A number of miRNAs showed reduced auxin sensitivity in the mutant compared with the wild type, consistent with the auxin-resistant phenotype of the mutant. Four miRNAs with significantly altered expression patterns in the mutant were further confirmed by Northern blot, which supported our microarray data. Clustering analysis revealed some novel auxin-sensitive miRNAs in roots. Analysis of miRNA duplication and expression patterns suggested the evolutionary conservation between miRNAs and protein-coding genes. MiRNA promoter analysis suggested the possibility that most plant miRNAs might share the similar transcriptional mechanisms with other non-plant eukaryotic genes transcribed by RNA polymerase II. Auxin response elements were proved to be more frequently present in auxin-related miRNA promoters. Comparative analysis of miRNA and protein-coding gene expression datasets uncovered many reciprocally expressed miRNA-target pairs, which could provide some hints for miRNA downstream analysis. Based on these findings, we also proposed a feedback circuit between miRNA(s) and ARF(s). The results presented here could serve as the basis for further in-depth studies of plant miRNAs involved in auxin signalling.
doi: 10.1007/s00425-009-0994-3 PubMed: 19655164 Google Scholar

05. Unveiling Long Non-coding RNA Networks from Single-Cell Omics Data Through Artificial Intelligence.
Cao G, Chen D*
Methods Mol Biol. 2025;2883:257-279. doi: 10.1007/978-1-0716-4290-0_11.
Single-cell omics technologies have revolutionized the study of long non-coding RNAs (lncRNAs), offering unprecedented resolution in elucidating their expression dynamics, cell-type specificity, and associated gene regulatory networks (GRNs). Concurrently, the integration of artificial intelligence (AI) methodologies has significantly advanced our understanding of lncRNA functions and its implications in disease pathogenesis. This chapter discusses the progress in single-cell omics data analysis, emphasizing its pivotal role in unraveling the molecular mechanisms underlying cellular heterogeneity and the associated regulatory networks involving lncRNAs. Additionally, we provide a summary of single-cell omics resources and AI models for constructing single-cell gene regulatory networks (scGRNs). Finally, we explore the challenges and prospects of exploring scGRNs in the context of lncRNA biology.
doi: 10.1007/978-1-0716-4290-0_11 PubMed: 39702712 Google Scholar

04. The ChIP-Hub Resource: Toward plantEncode.
Lan Y#, Zhao X#, Chen D*
Methods Mol Biol. 2023;2698:221-231. doi: 10.1007/978-1-0716-3354-0_14.
Recent advances in sequencing technologies lead to the generation of an enormous amount of regulome and epigenome data in a variety of plant species. However, a comprehensive standardized resource is so far not available. In this chapter, we present ChIP-Hub, an integrative platform that has been developed based on the ENCODE standards by collecting and reanalyzing regulatory genomic datasets from 41 plant species. The ChIP-hub website is introduced in this chapter, including information on detailed steps of searching, data download, and online analyses, which facilitates users to explore ChIP-Hub. We also provide a cross-species comparison of chromatin accessibility information that gives a thorough view of evolutionary regulatory networks in plants.
doi: 10.1007/978-1-0716-3354-0_14 PubMed: 37682478 Google Scholar

03. Integration of Genome-Wide TF Binding and Gene Expression Data to Characterize Gene Regulatory Networks in Plant Development.
Chen D*, Kaufmann K
Methods Mol Biol. 2017;1629:239-269. doi: 10.1007/978-1-4939-7125-1_16.
Key transcription factors (TFs) controlling the morphogenesis of flowers and leaves have been identified in the model plant Arabidopsis thaliana. Recent genome-wide approaches based on chromatin immunoprecipitation (ChIP) followed by high-throughput DNA sequencing (ChIP-seq) enable systematic identification of genome-wide TF binding sites (TFBSs) of these regulators. Here, we describe a computational pipeline for analyzing ChIP-seq data to identify TFBSs and to characterize gene regulatory networks (GRNs) with applications to the regulatory studies of flower development. In particular, we provide step-by-step instructions on how to download, analyze, visualize, and integrate genome-wide data in order to construct GRNs for beginners of bioinformatics. The practical guide presented here is ready to apply to other similar ChIP-seq datasets to characterize GRNs of interest.
doi: 10.1007/978-1-4939-7125-1_16 PubMed: 28623590 Google Scholar
02. From Genes to Networks: Characterizing Gene-Regulatory Interactions in Plants.
Kaufmann K, Chen D
Methods Mol Biol. 2017;1629:1-11. doi: 10.1007/978-1-4939-7125-1_1.
Plants, like other eukaryotes, have evolved complex mechanisms to coordinate gene expression during development, environmental response, and cellular homeostasis. Transcription factors (TFs), accompanied by basic cofactors and posttranscriptional regulators, are key players in gene-regulatory networks (GRNs). The coordinated control of gene activity is achieved by the interplay of these factors and by physical interactions between TFs and DNA. Here, we will briefly outline recent technological progress made to elucidate GRNs in plants. We will focus on techniques that allow us to characterize physical interactions in GRNs in plants and to analyze their regulatory consequences. Targeted manipulation allows us to test the relevance of specific gene-regulatory interactions. The combination of genome-wide experimental approaches with mathematical modeling allows us to get deeper insights into key-regulatory interactions and combinatorial control of important processes in plants.
doi: 10.1007/978-1-4939-7125-1_1 PubMed: 28623575 Google Scholar

01. Bridging Genomics and Phenomics
Chen D, Chen M, Altmann T, Klukas C
Methods Mol Biol. 2014;1629:299-333. doi: 10.1007/978-3-642-41281-3_11.
Genomics and phenomics are two fundamentally important branches of biological sciences, and they stand at both ends of the multiple “omics” families. A central goal of current biology is to establish complete functional links between the genome and phenome, the so-called genotype–phenotype map. Recent advances in high-throughput and high-dimensional genotyping and phenotyping technologies enable us to uncover the casual networks inside the “black box” that lies between genotypes and phenotypes using the principles of genome-wide association studies (GWAS). Application of GWAS and analogous methodologies and incorporation of multiple omics data begin to unravel the contribution of genetic variation to phenotypic diversity. Integrating “omics” data at broad levels by using the systems-biology approach is paramount to further bridging the gaps between genomics and phenomics and eventually making accurate predictions of phenotypes based on genetic contribution.