Genome-wide comparison of microRNAs and their targeted transcripts among leaf, flower and fruit of sweet orange.
Liu Y, Wang L, Chen D, Wu X, Huang D, Chen L, Li L, Deng X, Xu Q
BMC Genomics. 2014 Aug 20;15:695. doi: 10.1186/1471-2164-15-695.
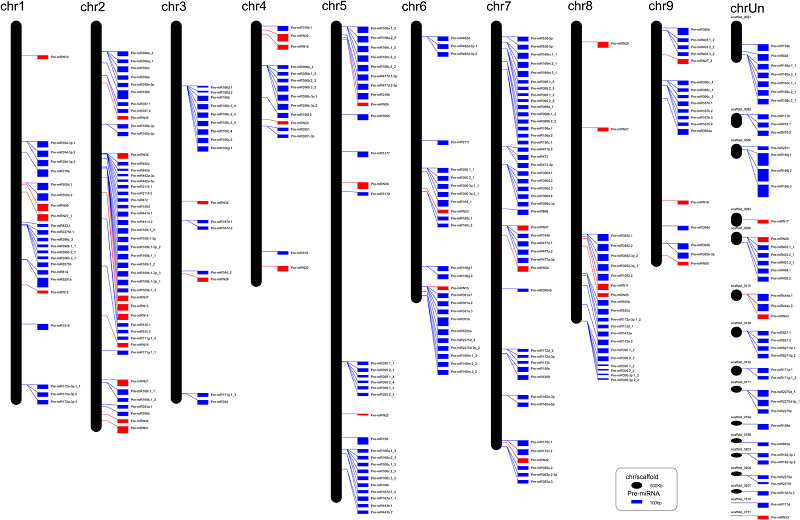
BACKGROUND: In plants, microRNAs (miRNAs) regulate gene expression mainly at the post-transcriptional level. Previous studies have demonstrated that miRNA-mediated gene silencing pathways play vital roles in plant development. Here, we used a high-throughput sequencing approach to characterize the miRNAs and their targeted transcripts in the leaf, flower and fruit of sweet orange. RESULTS: A total of 183 known miRNAs and 38 novel miRNAs were identified. An in-house script was used to identify all potential secondary siRNAs derived from miRNA-targeted transcripts using sRNA and degradome sequencing data. Genome mapping revealed that these miRNAs were evenly distributed across the genome with several small clusters, and 69 pre-miRNAs were co-localized with simple sequence repeats (SSRs). Noticeably, the loop size of pre-miR396c was influenced by the repeat number of CUU unit. The expression pattern of miRNAs among different tissues and developmental stages were further investigated by both qRT-PCR and RNA gel blotting. Interestingly, Csi-miR164 was highly expressed in fruit ripening stage, and was validated to target a NAC transcription factor. This study depicts a global picture of miRNAs and their target genes in the genome of sweet orange, and focused on the comparison among leaf, flower and fruit tissues. CONCLUSIONS: This study provides a global view of miRNAs and their target genes in different tissue of sweet orange, and focused on the identification of miRNA involved in the regulation of fruit ripening. The results of this study lay a foundation for unraveling key regulators of orange fruit development and ripening on post-transcriptional level.